Hypervitaminosis can occur when pharmaceutical vitamin D is taken in excess, with a wide variety of symptoms and signs related to hypercalciuria, hypercalcemia, and metastatic calcifications (Table 58-2). The toxic dosage has not been established for all ages, but infants and children are more susceptible. Toxicity should always be monitored when daily doses markedly exceeding the present upper limit of more than 50 µg are given for a longer period. Overproduction of renal 1,25(OH)2D by abnormal hormonal stimuli (as seen in fibroblast growth factor-23 [FGF-23] or Klotho-null mice) or absence of CYP24A1 (see later), the main catabolizing enzyme, causes the same calcemic side effects, with severe multiple-organ calcification (especially kidney, vascular wall, and heart valves) leading to premature death.24
Table 58-2. Symptoms of Vitamin D Toxicity
| Hypercalciuria |
| Kidney stones |
| Hypercalcemia |
| Hyperphosphatemia |
| Polyuria |
| Polydipsia |
| Decalcification of bone |
| Ectopic calcification of soft tissues (kidney and lung) |
| Nausea and vomiting |
| Anorexia |
| Constipation |
| Headache |
| Hypertension |
Most vertebrates also accomplish their needs for vitamin D by photochemical synthesis in the skin; therefore, vitamin D is not a true vitamin. It is formed from 7-dehydrocholesterol (7DHC or provitamin D3), which is present in large amounts in cell membranes of keratinocytes of the basal or spinous epidermal layers. By the action of ultraviolet B (UVB) light (290 to 315 mm), the B ring of 7DHC can be broken to form previtamin D3. Previtamin D3 is unstable, and in the lipid bilayer of membranes, it is rapidly isomerized to vitamin D3 by thermal energy, followed by transport to the serum vitamin D–binding protein and uptake into the liver for further metabolization.
The production of previtamin D3 is a nonenzymatic photochemical reaction which is not subject to regulation other than substrate (7DHC) availability and intensity of UVB irradiation. 7DHC is the last precursor in the de novo biosynthesis of cholesterol. The enzyme 7HDC-Δ7-reductase (or sterol Δ7-reductase) catalyses the production of cholesterol from 7DHC. Inactivating mutations of the 7DHC-Δ7-reductase gene25 are the hallmark of the autosomal recessive Smith-Lemli-Opitz syndrome, characterized by high tissue and serum 7DHC levels and multiple anomalies, including craniofacial dysmorphism and mental retardation due to the lack of cholesterol synthesis.26 These patients may exhibit sometimes increased serum vitamin D and 25(OH)D concentrations.27 Likewise, animals pretreated with a specific sterol-Δ7-reductase inhibitor also exhibit an augmented vitamin D synthesis following UVB irradiation.28 With increasing human age, cutaneous stores of provitamin D decrease, together with decreased photoproduction of vitamin D.16 In cats and the feline species in general, the high cutaneous sterol-Δ7-reductase activity hampers photoproduction of vitamin D, making it a true vitamin.29 Apart from substrate (7DHC) availability, the photochemical synthesis of vitamin D3 in the skin largely depends on the amount of UVB photons that strike the basal epidermal layers. Glass, sunscreen, clothes, and skin pigment absorb UVB and blunt vitamin D3 synthesis. Latitude, time of day, and season are factors that influence the intensity of solar radiation and the cutaneous production of vitamin D3. Therefore, there is a risk for a shortage of vitamin D supply during winter and spring. In both the Northern and Southern hemispheres above 40 degrees latitude, vitamin D3 synthesis of the skin decreases or disappears during winter months, owing to the low inclination of the sun and the atmospheric filtration of the shortest (but effective for vitamin D3 synthesis) UV waves of sunlight. The importance of skin synthesis of vitamin D3 to maintain normal vitamin D status is best reflected by the vitamin D deficiency observed in submarine personnel or inhabitants of Antarctica30 during prolonged absence of sun exposure, and also by the extremely high prevalence of vitamin D deficiency in countries where exposure to sunlight is extremely low for cultural and religious reasons, as in several Arabian countries with strict adherence to Islamic rules for body covering.31–34 Solar exposure of 2 hours per week of the face and hands is probably sufficient for maintaining normal 25(OH)D concentrations in children35 and adults but should be further fine-tuned according to the climate and latitude.36
Nature has built in several feedback mechanisms to minimize the risk that prolonged sun exposure would cause vitamin D intoxication. Cutaneous vitamin D and especially previtamin D are photosensitive and will be degraded to inactive sterols (lumisterol, tachysterol) before they are translocated to the circulation (Fig. 58-1). Only a maximum of 10% to 15% of the provitamin D will be converted to vitamin D. Sunlight-induced melanin synthesis, acting as a natural sunscreen, provides an additional negative feedback.
Metabolism of Vitamin D
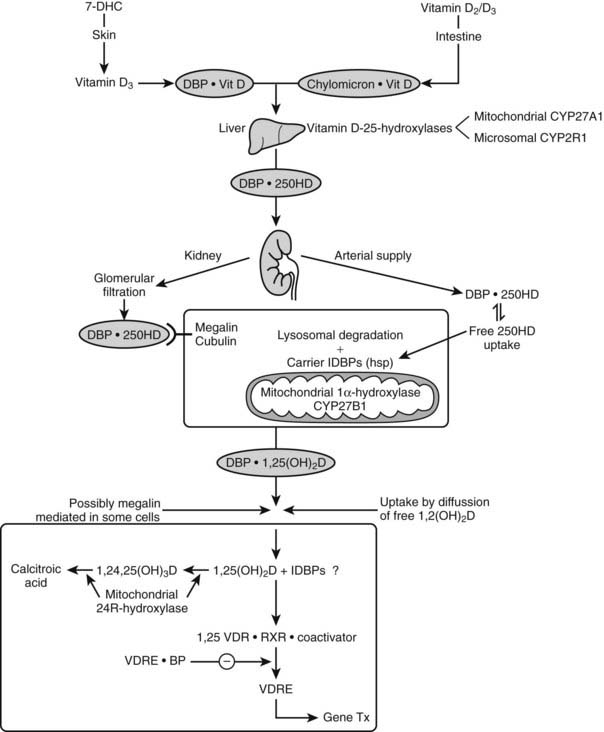
Vitamin D is biologically inert and requires two successive hydroxylations in the liver (on C25) and kidney (on the α position of C1), using cytochrome P450 enzymes37,38 to form its hormonally active metabolite, 1α,25-dihydroxyvitamin D (see Fig. 58-1).
25-HYDROXYLATION
25(OH)D was the first metabolite identified after the availability of radiolabeled vitamin D3.39,40 Although the liver is probably the main tissue responsible for 25-hydroxylation of vitamin D, extrahepatic 25-hydroxylation has been observed in vitro in a large number of tissues. In vivo observations after hepatectomy in rats also revealed that the conversion rate of [3H]-vitamin D was still about 10% when compared with intact rats.40 The hepatic 25-hydroxylation step is probably performed by more than one enzyme, localized either in the inner mitochondrial membrane (CYP27A1 or sterol 27-hydroxylase) or in the microsomes (including CYP2D11, CYP2D25, CYP3A4, and especially CYP2R1).37,41,42 CYP27A1 is a multifunctional enzyme with broad substrate specificity and is mainly involved in the 26- or 27-hydroxylation of cholesterol and bile-acid precursors.37 The rather mild (if any) disturbance of vitamin D metabolism in animals or humans lacking this enzyme43 indicates that the 25-hydroxylation of vitamin D does not rely exclusively on the activity of CYP27A1. It is therefore likely that a microsomal enzyme is the more physiologic enzyme, as initially suspected on the basis of hepatic enzyme activity being much higher and with lower Km in the microsomal fraction when compared with mitochondrial 25-hydroxylase activity.44 The microsomal enzyme activity is up-regulated by vitamin D deficiency or by prior exposure to phenobarbital. The most important 25-hydroxylase is probably CYP2R1, since a homozygous mutation was identified in a patient with classical rickets with low 25(OH)D levels.42
1α-HYDROXYLATION
25(OH)D is biologically inactive and requires further hydroxylation in the kidney45,46 to the active hormone, 1,25(OH)2D, by 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1). The production of 1,25(OH)2D is regulated primarily at this final step by several factors (vide infra). The rat, mouse, and human 1α-hydroxylase have been cloned by several groups47–51 and mapped on human chromosome 12q13.3 in close vicinity to the VDR gene. The proximal renal tubule is the principle site of 1α-hydroxylation, but high levels of 1α-hydroxylase mRNA have also been found in human keratinocytes,48 and its gene expression is also observed in mouse macrophages52 and about ten other tissues.24 1α-Hydroxylase activity is under tight control by 1,25(OH)2D (negative but probably indirect feedback); parathyroid hormone (PTH), calcitonin, and insulin-like growth factor 1 (all positive feedback); and phosphate, calcium, and especially FGF-23 (negative regulation).38,53,54 The promoters of the mouse and human 1α-hydroxylase genes have been characterized with a profound responsiveness to PTH and a negative regulation by 1,25(OH)2D55,56 by complex chromatin and DNA modifications.57
Pseudovitamin D–deficiency rickets (PDDR), also known as vitamin D-dependency rickets type I, is an autosomal recessive disease characterized by failure to thrive, muscle weakness, skeletal deformities, hypocalcemia, secondary hyperparathyroidism, normal to high serum levels of 25(OH)D, and low serum 1,25(OH)2D concentrations, all caused by impaired activity of the renal 1α-hydroxylase.58 These patients recover with supplementation of physiologic doses of 1,25(OH)2D. The human CYP27B1 maps to the previously identified PDDR locus and mutations found in this gene in patients with PDDR provide the molecular genetic basis for the disease.48,59
24-HYDROXYLATION: CATABOLISM OR SPECIFIC FUNCTION?
An alternative hydroxylation of 25(OH)D occurs on carbon 24 by the multifunctional enzyme, 24-hydroxylase (CYP24A), mapped on human chromosome 20q13.60 This enzyme not only initiates the catabolic cascade of 25(OH)D and 1,25(OH)2D61,62 by 24-hydroxylation but catalyzes also the dehydrogenation of the 24-OH group and performs 23-hydroxylation, resulting in 24-oxo-1,23,25(OH)3D.62 This C24 oxidation pathway finally leads to calcitroic acid, which is the major end product of 1,25(OH)2D (Fig. 58-2). In vivo evidence for this catabolic role of 24-hydroxylase was provided by the generation of mice deficient in the 24-hydroxylase gene, resulting in pathology consistent with systemic excess of 1,25(OH)2D.63 The expression of the 24-hydroxylase gene has been detected in virtually all nucleated cells. The induction of CYP24A belongs to the most sensitive biomarkers for responsiveness and is explained by the presence of several vitamin D responsive elements in its promoter.64,65 As a consequence, CYP24A mRNA levels appear to fall under the detection limits in VDR knockout mice.50,66 No human mutation in the 24-hydroxylase gene have yet been identified, but local overexpression of the enzyme may be involved in cancer.67
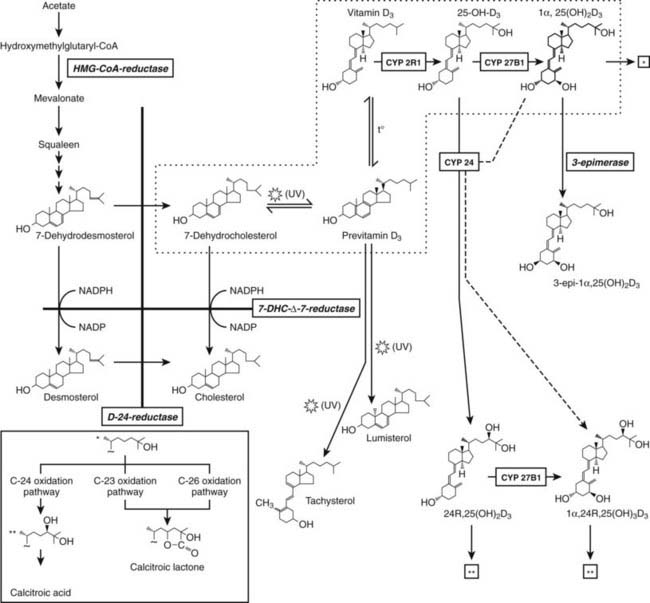
OTHER METABOLIC PATHWAYS FOR VITAMIN D AND ITS METABOLITES
Apart from the multifunctional 24-hydroxylation pathway, C23 and C26 hydroxylation of 1,25(OH)2D is also possible in the absence of prior 24-hydroxylation. The 23-hydroxylation probably only becomes important in the case of vitamin D excess; its major locus is the kidney. In contrast, 26-hydroxylation is mainly performed outside the kidney. Both activities are necessary for the formation of 25(OH)D- or 1,25(OH)2D-23,26-lactones (see Fig. 58-1). The A-ring metabolism involves the oxidation of C19 and the recently discovered 3-epimerisation. The latter, irreversible, reaction occurs only in a limited number of cells (e.g., keratinocytes, bone, and parathyroid cells) and is performed by hydroxysteroid dehydrogenases.68
The enzymes involved in the metabolic degradation of 1,25(OH)2D do not recognize all vitamin D analogs in the same way. Indeed, analogs with either 20-epi or 20-methyl configuration or 16-ene structure show an impaired 23-hydroxylation. These or other analogs are then preferentially hydroxylated on C26 or on new terminal carbons of the side chain. Such alternative metabolism can certainly explain part of the specific selectivity profile of a number of analogs (vide infra).
Vitamin D is mainly excreted in the bile after esterification in the liver, but some of its more polar metabolites (e.g., calcitroic acid) are excreted via the urine. The enterohepatic recirculation of vitamin D esters is probably devoid of biological relevance.
Vitamin D Transport
Nutritional vitamin D is absorbed by the gut and then transported via the lymphatic system by chylomicrons69 and stored in several tissues (e.g., fat and muscle). Skin-produced vitamin D probably binds directly to an α-globulin known as vitamin D binding protein (DBP) and is then transported to the liver, where it is hydroxylated and thereafter released as 25(OH)D.
Human DBP,70 detected immunologically in 1959 as a group-specific component, or Gc-globulin,71 is a 458-amino-acid glycoprotein synthesized by the liver. Long before DBP’s functions had been characterized, its polymorphicity was already used in population genetics, parentage testing, and forensic medicine.72 Worldwide, over 120 Gc alleles have been detected,73 making the DBP locus one of the most polymorphic known. The Gc1F, Gc1S and Gc2 are the three most common alleles. Since in the many thousands of sera tested, none had been found of DBP deficiency, such a mutation was for a long time considered to be lethal; but this was contradicted by the generation of viable and fertile homozygous, DBP-deficient mice (DBP-null animals).74 The existence of similarity among the genes and protein structure of DBP, albumin, and α-fetoprotein is long recognized.75 The crystal structure, with or without actin, is now available and identified a surface cleft to bind 25(OH)D.76
ROLE OF VITAMIN D BINDING PROTEIN FOR VITAMIN D HOMEOSTASIS
DBP, the major plasma carrier of vitamin D3, all its metabolites, and the vitamin D3 analogs, has one vitamin D sterol-specific binding site.75 The relative binding affinity is 25(OH)D-23,26-lactone > 25(OH)D = 24,25(OH)2D = 25,26(OH)2D (Ka = 5.108 mol/L at 4° C for human DBP) 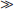 1,25(OH)2D (4.107 mol/L)
1,25(OH)2D (4.107 mol/L) 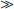 vitamin D
vitamin D 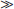 previtamin D.77 The affinity for D2 metabolites is slightly lower than for D3 metabolites in mammals, but especially in birds. Since probably only non-DBP-bound vitamin D metabolites can readily cross the plasma membrane, and since the VDR has a much higher affinity for 1,25(OH)2D than for 25(OH)D (100-fold difference), while the opposite is true for DBP, it is clear that 1,25(OH)2D has substantially higher cellular uptake than 25(OH)D. This is also confirmed by the distribution space of (radiolabeled) metabolites: 25(OH)D has a distribution space similar to that of DBP and the plasma volume, whereas the distribution space of 1,25(OH)2D is closer to that of intracellular water. The half-life of 25(OH)D and 1,25(OH)2D in the human circulation is about 2 to 3 weeks and 4 to 6 hours, respectively.78,79 DBP’s function in the vitamin D endocrine system is assumed to reflect the “free hormone” hypothesis, which states that the unbound (free) rather than the protein-bound fraction of the active vitamin D hormone is responsible for the biological activity. The plasma concentration of DBP is increased by estrogens in most mammalian species and birds. In women, the DBP concentration therefore doubles at the end of pregnancy.80 Recent studies with megalin knockout mice indicate that megalin, a lipoprotein-like receptor present at the surface of the proximal tubular cells in the kidney, is responsible for the reabsorption of DBP and of DBP complexed with vitamin D sterols. This megalin reabsorption mechanism may control the availability of the 25(OH)D/DBP complex for the 25(OH)D-1α-hydroxylation enzyme and explain the severe bone disease of megalin-deficient mice.81
previtamin D.77 The affinity for D2 metabolites is slightly lower than for D3 metabolites in mammals, but especially in birds. Since probably only non-DBP-bound vitamin D metabolites can readily cross the plasma membrane, and since the VDR has a much higher affinity for 1,25(OH)2D than for 25(OH)D (100-fold difference), while the opposite is true for DBP, it is clear that 1,25(OH)2D has substantially higher cellular uptake than 25(OH)D. This is also confirmed by the distribution space of (radiolabeled) metabolites: 25(OH)D has a distribution space similar to that of DBP and the plasma volume, whereas the distribution space of 1,25(OH)2D is closer to that of intracellular water. The half-life of 25(OH)D and 1,25(OH)2D in the human circulation is about 2 to 3 weeks and 4 to 6 hours, respectively.78,79 DBP’s function in the vitamin D endocrine system is assumed to reflect the “free hormone” hypothesis, which states that the unbound (free) rather than the protein-bound fraction of the active vitamin D hormone is responsible for the biological activity. The plasma concentration of DBP is increased by estrogens in most mammalian species and birds. In women, the DBP concentration therefore doubles at the end of pregnancy.80 Recent studies with megalin knockout mice indicate that megalin, a lipoprotein-like receptor present at the surface of the proximal tubular cells in the kidney, is responsible for the reabsorption of DBP and of DBP complexed with vitamin D sterols. This megalin reabsorption mechanism may control the availability of the 25(OH)D/DBP complex for the 25(OH)D-1α-hydroxylation enzyme and explain the severe bone disease of megalin-deficient mice.81
OTHER FUNCTIONS OF VITAMIN D BINDING PROTEIN
DBP binds globular actin with a high affinity (Ka = 2 × 109 mmol/L).82,83 Actin is the most abundant intracellular protein. The cell motility, shape, and size depend on the ability of globular actin to polymerize into filaments (F-actin). Upon cell injury or cell necrosis, actin is released into extracellular space. However, when actin is released from cells, it may rapidly form filaments with detrimental effects for the microcirculation. Two plasma proteins, DBP and gelsolin, bind actin avidly, thereby acting as “actin-scavenger” system.84,85
DBP-null (KO) mice, however, develop normally. They are nevertheless more sensitive to vitamin D deficiency and less sensitive to vitamin D excess, probably by an enhanced urinary loss of vitamin D metabolites.74 The DBP and megalin KO mice, however, suggest that the main function of DBP is indeed to transport all vitamin D metabolites and preserve them from rapid clearance or urinary loss.
Action and Mode of Action
GENERAL CHARACTERISTICS OF THE VITAMIN D RECEPTOR
Protein
1,25(OH)2D, the hormonally active form of vitamin D, exerts its effects mainly by activating the nuclear VDR, a member of the nuclear-receptor superfamily of ligand-activated transcription factors. Based on structure and function similarities between members of this family, different functional domains can be distinguished in these nuclear-receptor proteins. The short A/B domain at the N-terminus of VDR lacks the usual ligand-independent activation function (AF1). Two highly conserved zinc finger DNA binding motifs constitute the DNA-binding C domain, which also harbors the nuclear localization signal. The D domain or hinge region may regulate the receptor’s flexibility between DNA-binding and ligand-binding domains and may be crucial to allowing the heterodimer complex of the ligand-binding domains to interact with two differently oriented response elements (direct repeat or palindrome orientation with variable number of spacer nucleotides). The large multifunctional E region contains the ligand-binding domain, as well as a dimerization surface and a ligand-dependent activation function (AF2) at the extreme C-terminus, represented by helix 12.86
Gene
The human VDR gene, consisting of 14 exons, spans more than 60 kb on chromosome 12.87,88 The major VDR transcript is a 4.8 kb mRNA species, but multiple promoters and alternative splicing give rise to a multitude of less abundant transcripts that mostly vary in their 5′ untranslated region but encode the same 427-amino-acid protein.88 However, two of these mRNAs are translated into VDR proteins that contain an additional 23 or 50 amino acids at the N-terminus.88
Genomic Actions
Binding of 1,25(OH)2D to VDR generates conformational changes of VDR followed by heterodimerization with unliganded RXR and binding to vitamin D response elements (VDREs) in the promoter region of vitamin D target genes, with subsequent release of corepressors and recruitment of coactivators and general transcription factors for the assembly of an active transcriptional complex.89 A putative crucial event in this respect is the mousetrap-like intramolecular folding of helix 12, closing off the ligand-binding pocket and exposing the AF2 domain for interaction with coactivators.90 Corepressors bind and silence unliganded steroid receptors by recruitment of histone deacetylases, maintaining chromatin in a transcriptional repressive state.91 Coactivators are a group of proteins that allow gene transcription in several waves of activities. First, coactivators of the CBP/p300 family and of the p160 protein family, including the steroid receptor coactivators (SRCs), are recruited.92–94 These proteins possess intrinsic histone acetyltransferase (HAT) activity and by acetylating histone tails open up the chromatin structure, creating a chromatin environment permissive for gene transcription.95 In a second wave, the vitamin D receptor interacting protein (DRIP) multimeric complex is recruited, followed by recruitment of basal transcription factors, as well as RNA polymerase II. Finally, target gene transcription is induced.96 Gene expression can also be mediated by ATP-dependent chromatin remodeling complexes such as SWI/SNF-type and ISWI-type complexes and the multiprotein complex WINAC.97–99 Distinct regulation of transcriptional coregulators may provide species-specific, tissue-specific, or developmental stage–specific regulation of nuclear receptor function.100 Furthermore, the expression or the recruitment of these regulatory proteins is regulated by several intracellular signaling pathways101 and by steroids themselves,100 with receptor agonists or antagonists inducing preferential recruitment of coactivators or corepressors, respectively.101
A hexanucleotide direct repeat spaced by three nucleotides (DR3) is the cognate vitamin D response element (VDRE) to which RXR and VDR bind the 5′ and 3′ half-site, respectively, although alternative options appear to be possible, both with respect to dimer formation (VDR/VDR, VDR/RAR) and target-gene VDRE structure (DR4, DR6; IP9).102
Nongenomic Actions
Aside from the VDR transcriptional or genomic effects, several research groups have described rapid effects by 1,25(OH)2D that are independent of transcription and would be mediated by a membrane receptor for 1,25(OH)2D or by the localization of the nuclear VDR near the membrane.103 These so-called nongenomic effects include the opening of calcium or chloride channels and the activation of second messenger signaling pathways (phosphoinositide turnover, activation of protein kinase C, and the Ras/Raf/ERK/MAPK pathway). A wide variety of rapid and transient modifications in the second messenger signaling system have also been observed for other steroid hormones.104 At the tissue or cellular level, however, nongenomic activity of vitamin D and its analogs or metabolites have only been described for intestinal calcium absorption (transcaltachia) or cellular differentiation of leukemia cells.105–107 This pathway seems to prefer 6-s-cis to the 6-s-trans configuration of vitamin D.105 Moreover, the agonist/antagonist specificity differs for that of the genomic pathway.108
CLASSIC TARGET TISSUES
The action of 1,25(OH)2D on bone, intestine, kidney, and parathyroid glands and its role in mineral metabolism is the result of a complex interplay between calcium and phosphate 1,25(OH)2D, PTH, and phosphatonins. PTH induces calcium mobilization from bone and stimulates 1,25(OH)2D production, but its secretion is inhibited by the action of 1,25(OH)2D on the parathyroid glands (negative feedback). In a second negative feedback loop, 1,25(OH)2D limits its own availability by inhibition of 1α-hydroxylase and stimulation of 24-hydroxylase, inducing 1,25(OH)2D catabolism. In the last few years, considerable progress has been made in the understanding of phosphate homeostasis.109 The phosphaturic hormone phosphatonin, or FGF-23, is produced by osteocytes and osteoblasts and inhibits the activity of the NPT2 protein. The NPT2 gene encodes a renal sodium/phosphate cotransporter responsible for reabsorption of phosphate and represents a newly identified target gene for 1,25(OH)2D.110 Phosphatonin can be indirectly inactivated by a protease encoded by the PEX gene, which was identified as the gene that is defective in X-linked hypophosphatemic rickets. FGF-23 secretion is stimulated by 1,25(OH)2D and impairs renal 1α-hydroxylase, creating an additional feedback system so that the production of 1,25(OH)2D is tightly feedback regulated (Fig. 58-3).
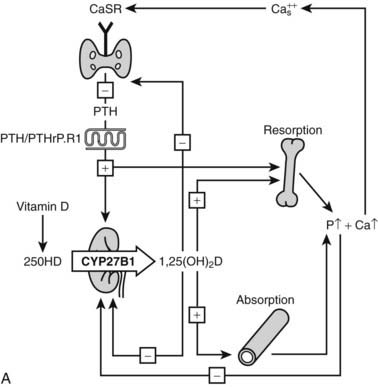
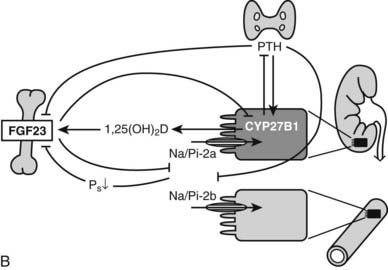
FIGURE 58-3. Feedback regulation of renal synthesis of 1,25(OH)2D. A, Regulation of renal 1,25(OH)2D synthesis by parathyroid hormone and calcium, with multiple feedback control mechanisms. CaSR, Calcium-sensing receptor. B, Regulation of renal 1,25(OH)2D synthesis by FGF23 and phosphate, with several feedback control mechanisms.
Effects on Intestine
The absorption capacity of calcium along the gastrointestinal tract of the rat is dependent on the segment and follows the order ileum > jejunum > duodenum. The efficiency of the small intestine to absorb dietary calcium is increased by 1,25(OH)2D,111 and the abundance of the vitamin D receptor is highest in the duodenum, followed by jejunum and ileum. Although the exact mechanism by which 1,25(OH)2D alters the flux of calcium across the intestinal absorptive cell is not known, 1,25(OH)2D increases the production and activity of several proteins in the small intestine, including TRPV6 and V5, calbindin-D9K, alkaline phosphatase, and low-affinity Ca-ATPase (PMCA). The entry of Ca2+ from the intestinal lumen across the brush border membrane into the enterocyte is mainly regulated by the epithelial channels TRPV6 and V5.112 The intracellular calcium transfer is considered to be dependent mainly on calbindin-D9K.113 The transfer of Ca2+ from the cytoplasm to the extracellular space requires energy input because of an uphill concentration gradient and an unfavorable electrochemical gradient. Both the plasma membrane calcium pump and a sodium-calcium exchanger play important roles in this process. The stimulatory effect of 1,25(OH)2D on the ATP-dependent uptake of Ca2+ at the basolateral membrane involves an increase in PMCA gene expression.114 The essential role of the intestine for calcium and phosphate homeostasis was clearly demonstrated by the phenotype of VDR KO mice. Such VDR-null mice are phenotypically normal at birth, but after weaning, they develop hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia despite very high levels of 1,25(OH)2D. They become growth retarded and develop severe rickets.66,112,115,116 Mice deficient in 1α-hydroxylase display a similar phenotype.117,118 This bone and calcium phenotype can be largely corrected by a high dietary calcium intake (especially in combination with high lactose intake) in both knockout models or 1,25(OH)2D treatment of 1α hydroxylase–null mice.112,119–125 These findings confirm previous observations in humans.59,126,127 The data strongly suggest that the intestine is the primary target for 1,25(OH)2D’s action on calcium/bone homeostasis. This is largely confirmed by genetic mouse models of selective rescue or deletion of VDR in the intestine of transgenic mice.128 The primary molecular targets, however, merit further exploration; ablation of CaBP-9k or TRPV6 or even their combined deficiency have shown no major effects on basal intestinal calcium absorption or serum calcium levels when calcium intake is normal.129 Paracellular intestinal calcium transport may also be part of the picture of vitamin D’s action in that the expression of claudin 2 and claudin 12, both known to form paracellular calcium channels, are induced by 1,25(OH)2D and decreased in the intestine of VDR-null mice.130
Effects on Kidney
The kidney is important both for the metabolism of 1,25(OH)2D and the reabsorption of calcium and phosphate, processes regulated by 1,25(OH)2D. The kidney and more specifically the proximal tubule is the central tissue for 1α-hydroxylation of 25(OH)D. Chronic renal failure reduces 1α-hydroxylase activity, which ultimately results in renal osteodystrophy or uremic bone disease. 1,25(OH)2D also increased the distal tubular reabsorption of calcium; as in the intestine, TRP channels (now TRPV5), calbindin-D9K and 28K, and the plasma membrane calcium ATPase are involved. Whereas in the intestine, active calcium absorption in the duodenum takes place before the less-regulated diffusion process in the ileum, reabsorption of filtered calcium follows a more logical sequence of massive calcium-sodium reabsorption in the proximal convoluted tubuli, followed by specific, actively regulated calcium reabsorption in the distal parts of the nephron. The crucial role of 1,25(OH)2D-regulated renal calcium reabsorption was demonstrated by persistent hypercalciuria and reduction in bone mass in TRPV5-deficient mice.131 The kidney is also the major component in phosphate homeostasis, as both PTH and FGF-23, in complex interplay with 1,25(OH)2D, are able to reduce renal phosphate reabsorption (see Fig. 58-3).
Effects on Bone
1,25(OH)2D has dual effects on bone: it can stimulate osteoclastogenesis and bone resorption as well as modify osteoblast function and bone mineralization. The overall effects of vitamin D metabolites on bone are thus extremely complex. From observations in man and animals, it is clear that vitamin D deficiency or resistance impairs bone-matrix mineralization, whereas osteoblast activity and matrix synthesis are even stimulated. Excess 1,25(OH)2D can clearly enhance osteoclastogenesis and bone resorption (Fig. 58-4). Because bone mineralization and bone structure can be largely normalized in vitamin D- or 1,25(OH)2D-deficient or resistant mice by sufficient supply of minerals via active or passive intestinal calcium absorption, it seems that direct effects of vitamin D metabolites on chondrocytes and bone cells are redundant if calcium and phosphate supply are guaranteed. However, most of the genes and proteins typically expressed in osteoblasts and osteoclasts are vitamin D regulated, so it is likely that 1,25(OH)2D can fine-tune bone mineral homeostasis. Moreover, pharmacologic use of vitamin D metabolites or analogs might positively influence bone balance, as shown by human and animal experiments132,133 and transgenic mice overexpressing osteoblast VDR.134
FIGURE 58-4. Effect of 1,25(OH)2D on bone cells and osteoclastogenesis. In osteoblast/stromal cells, 1,25(OH)2D induces ODF expression, down-regulates OPG, and stimulates M-CSF production. It also stimulates production of IL-6 and IL-11, which represent distinct signals in osteoclastogenesis. On osteoclast precursors, 1,25(OH)2D induces the expression of RANK (or ODF receptor) and several osteoclast differentiation markers, such as the vitronectin receptor αvβ3 and carbonic anhydrase-II. CA-II, Carbonic anhydrase-II; c-fms, M-CSF receptor; M-CSF, macrophage colony-stimulating factor; ODF, osteoclast differentiation factor; OPG, osteoprotegerin; V-ATP-ase, vacuolar adenosine triphosphatase.
Effects on Growth Plate
The absence of VDR or 1α-hydroxylase creates no detectable phenotype in overall growth or growth plate of prenatal animals, but the longitudinal growth of long bones is impaired after weaning. X-ray analysis reveals advanced rickets, including widening of the epiphyseal growth plate, with an increased width and marked disorganization of the growth plate on histology, including impaired mineralization of hypertrophic chondrocytes.66,115–118 This increased growth-plate width in VDR or 1α hydroxylase–null mice cannot be explained by their (normal) chondrocyte proliferation and differentiation, including collagen X and osteopontin expression. The expansion of the growth plate can be largely explained by decreased apoptosis of hypertrophic chondrocytes.135 Based on analysis of several genetic models with abnormal phosphate homeostasis, serum phosphate levels are probably crucial for hypertrophic chondrocyte apoptosis in vivo. This is confirmed in vitro: apoptosis of hypertrophic chondrocytes is regulated by phosphate levels via the activation of the caspase-9-mediated mitochondrial pathway.136
In accordance with these findings, chondrocyte-specific inactivation of the VDR did not cause a growth-plate phenotype and certainly not rickets.137 Critical analysis of these mice, however, revealed that VDR action in chondrocytes regulates bone development and phosphate homeostasis by inducing expression of paracrine factors such as vascular endothelial growth factor and receptor activator of nuclear factor κB (NFκB) ligand expression, leading to impaired vascular invasion and decreased osteoclast number in the metaphysic, as well as increased bone mass of long bones of juvenile chondrocyte-specific VDR-null mice. In addition, FGF-23 expression in osteoblasts was decreased, probably linked to the increased gene expression profile of NPT2 and 1α-hydroxylase in the kidney and resulting in increased serum levels of phosphate and 1,25(OH)2D.137
NONCALCEMIC OR NONCLASSIC ACTIONS OF VITAMIN D ENDOCRINE SYSTEM
The virtual ubiquitous expression of the VDR in all nucleated cells, the presence of a functional 1α-hydroxylase in at least 10 different tissues apart from the kidney, and the very large number of genes that are under direct or indirect control of 1,25(OH)2D all point toward a more universal role for the vitamin D endocrine system than just regulation of calcium/phosphate/bone metabolism. This is not totally unexpected; most other ligands for nuclear receptors also have a very wide spectrum of activities such as androgens, estrogens, glucocorticoids, and retinoids.24 Based on controlled observations in cells, tissues, and transgenic mice and on observational studies in humans, it seems that the functioning of nearly all major tissues or systems of the organism is modulated by vitamin D.
Skin
The combined presence of vitamin D production, 25-hydroxylase, 1α-hydroxylase, and VDR expression in the epidermis suggests the existence of a unique vitamin D intracrine system in which UVB-irradiated keratinocytes may supply their own needs for 1,25(OH)2D. A role for vitamin D in epidermal homeostasis can also be expected from the prominent effects of vitamin D compounds on keratinocyte growth and differentiation.138 The epidermal keratinocyte represents the major cell type in the epidermis and most likely the major cutaneous target cell for vitamin D, but many other cell types present in the epidermis are also vitamin D targets.
Based on studies of 1α hydroxylase–deficient mice, the repair of the essential barrier function of the skin is impaired in the absence of vitamin D action.139 The major skin phenotype of both VDR-null mice and children with VDR mutations is, however, the development of total alopecia. Hair development at birth is normal, but hair loss starts after the first catagen and ultimately leads to alopecia totalis associated with large dermal cysts. The absence of alopecia in vitamin D–deficient WT mice or in mice with CYP27B1 mutations clearly suggests that the absence of receptor and not its ligand is the cause of the skin phenotype. Mutations in the NR corepressor, hairless, or keratinocyte-specific loss of interaction of Lefl with β-catenin (part of the Wnt signaling pathway) produce a strikingly similar alopecia. There is therefore little doubt that ligand-independent effects of the VDR are required for normal keratinocyte stem cell function.
As expected from animal studies, no clear skin disorders are linked to vitamin D deficiency or insufficiency in humans. The very same photons that can generate the photoconversion of 7-dehydrocholesterol into previtamin D are also able to cause DNA damage and, ultimately, photoaging and increase the risk for skin cancer, so exposure to the UVB or sunlight needed to produce vitamin D always involves a small but cumulative risk of skin damage. This risk is especially relevant for humans with a fair skin type (phototypes 1 and 2).140 Although 1,25(OH)2D is able to generate a strong photoprotective effect against UVB-mediated events in cultured keratinocytes,141,142 the overall effect is negative.
Cell Proliferation and Cancer
Exposure to 1,25(OH)2D of virtually all normal cells and even most malignant cells results in an accumulation in the G0/G1 phase of the cell cycle.143–145 This inhibition of cell proliferation involves a large number of mechanisms and genes, and the exact sequence of events between VDR-mediated transactivation of genes and the actual G0/G1 arrest is probably cell-type specific. A general downstream effect is the regulation of the E2F family of transcription factors, which act as master switch for a very large number of genes involved in cell-cycle progression. These EF factors are under the control of the retinoblastoma protein members (especially pocket proteins p107 and p130), and their phosphorylation state is regulated by cyclins and cyclin-dependent kinases (p18, p19, p21, or p27), many of which are regulated by 1,25(OH)2D.143,146 However, 1,25(OH)2D may also inhibit cell growth by interfering with signaling pathways initiated by TGF-β, epidermal growth factor (IGF), prostaglandins,147 and Wnt ligands,148 as well as by intervening in other mitogenic signaling pathways (e.g., ERK/MAPK pathway and c-myc) (Fig. 58-5).149–156 Moreover, 1,25(OH)2D can regulate apoptosis and angiogenesis, mechanisms well known to be important for cancer cell expansion. In view of these well-established in vitro effects, one might expect a greater sensitivity for carcinogenesis in VDR-null mice. Epidermal, mammary, and intestinal cells of such animals do indeed show signs of hyperproliferation. Moreover, when exposed to chemocarcinogens or oncogens, VDR-null mice develop more mammary cancer–type lesions, skin tumors, and lymphomas.157,158
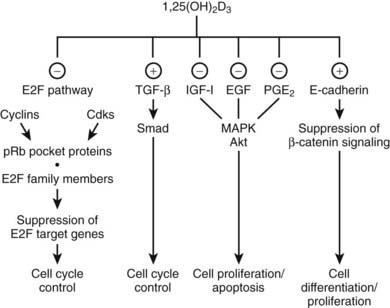
FIGURE 58-5. Effect of 1,25(OH)2D on cell-cycle progression. 1,25(OH)2D treatment leads to a cell cycle phase–specific effect characterized by an accumulation of cells in G1 through modulation of different signaling pathways. (−), Inhibitory effect; (+), stimulatory effect; EGF, epidermal growth factor; IGF-1, insulin-like growth factor 1; PGE2, prostaglandin E2; pRb, retinoblastoma tumor-suppressor gene; TGF-β, transforming growth factor β.
In humans, absolute VDR or CYP27B1 deficiencies are rare, but vitamin D deficiency is highly frequent. This obviously raises the question whether such vitamin D deficiency is associated with increased risk of cancer in humans. Such a hypothesis was originally reinforced by observations of higher cancer prevalence in areas of the United States and Japan with lower UVB exposure. Serum concentrations of 25(OH)D are, of course, a much better indication of the real vitamin D status, and a vast literature links lower levels of 25(OH)D with higher prevalence of the major cancers, especially colon and breast cancer, with more mixed results for prostate cancer. The inverse association between colorectal cancer or breast cancer and serum 25(OH)D levels was confirmed by the results of the Third National Health and Nutrition Examination Survey (NHANES III),159 although 25(OH)D levels were not related to overall cancer mortality. In most of the vast number of cross-sectional or observational studies, the higher cancer risk was found in subjects with serum 25(OH)D levels below 20 ng/mL, but most studies also revealed a significant trend across the different 25(OH)D subgroups and risk of cancer. Meta-analysis of studies addressing the association between 25(OH)D levels suggest that women with serum 25(OH)D of approximately 48 ng/mL (median of the top quintile) had a 50% lower risk of breast cancer than those with serum less than 13 ng/mL in the lowest quintile,160 and that individuals with serum 25(OH)D levels greater than 32 ng/mL had a 50% lower incidence of colorectal cancer than those with relatively low levels (≤12 ng/mL).161 However, a number of studies also link higher vitamin D nutritional status with a higher prevalence or more aggressive type of cancer.24,162
The final question is, of course, whether serum 25(OH)D is a predictor or has a causative relation with the overall cancer risk. Intervention studies should be able to provide the answer. In the Women’s Health Initiative (WHI) study, a significant inverse relationship was found between baseline levels of serum 25(OH)D and subsequent colorectal cancer incidence, but postmenopausal women receiving calcium (1 g) plus vitamin D (400 IU) did not develop less colon cancer than control patients.163 In a much smaller 4-yr study in postmenopausal women, higher doses of calcium (1.4 to 1.5 g) and vitamin D (1100 IU), which raised serum 25(OH)D to mean levels above 80 nmol/L, did significantly reduce overall cancer risk.164 The small number of cancer deaths and a major confounding factor of calcium intake, however, limit the value of this study to hypothesis, and much larger prospective studies with substantial vitamin D supplementation are essential.
Immune Function and Vitamin D
All immune cells (antigen-presenting cells, T and B cells, natural killer [NK] cells, and even mast cells) express at certain stages of their differentiation a functional VDR. Antigen-presenting cells (dendritic cells and equivalent resident cells, as well as monocytes/macrophages) can synthesize 1,25(OH)2D using the same enzyme as in the kidney but controlled by immune stimuli instead of calciotropic hormones.52,165–167 Finally, 1,25(OH)2D regulates a wide range of genes that play crucial roles in the immune system. The overall effects are different for the innate immune system (largely mediated by monocytes/macrophages) than for the acquired immune system. The innate immune system, upon exposure to bacterial agents, is first stimulated to produce 1,25(OH)2D (Fig. 58-6A) and thereafter activated by this paracrine 1,25(OH)2D to become a more active macrophage, including the local production of a number of defensins (including cathelicidin). Defects in immune functions indispensable for antimicrobial activity have been observed in vitamin D–deficient mice.168,169 The overall effects suggest that 1,25(OH)2D enhances the natural defense against bacterial infection. In the human situation, low serum levels of 25(OH)D were repeatedly associated with increased susceptibility to and more rapid disease progression of tuberculosis.170–174 This hypothesis is confirmed by the lower induction of cyclic adenosine monophosphate (cAMP) by monocytes incubated with 25(OH)D-deficient serum from sunlight-deprived African Americans.175 Moreover, oral administration of vitamin D markedly improved tuberculosis outcome in small-scale studies.176,177 Prospective clinical trials are ongoing to evaluate the effect of vitamin D supplementation on the evolution of various infections, including tuberculosis, to confirm the cause/effect relationship between vitamin D status and the native immune defense system.
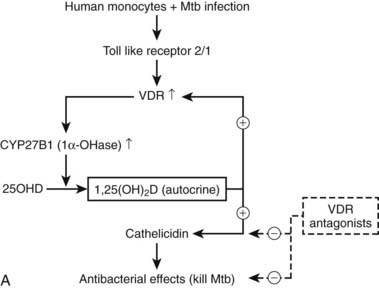
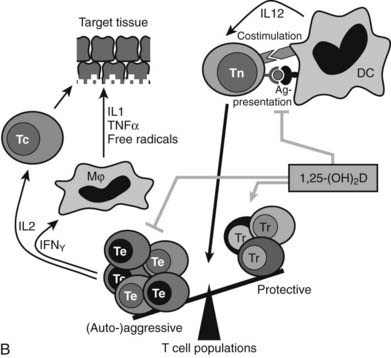
FIGURE 58-6. Immune effects of the vitamin D endocrine system. In the innate immune system, 1,25-(OH)2D strengthens the antimicrobial function of monocytes/macrophages, for example, through enhanced expression of the cathelicidin antimicrobial peptide, eventually leading to better clearance of pathogenic microorganisms. In the acquired immune system, the immunomodulatory effects of 1,25(OH)2D on players of the adaptive immune system can lead to the protection of target tissues in autoimmune diseases and transplantation. 1,25(OH)2D inhibits the surface expression of MHC II–complexed antigen and of costimulatory molecules, as well as the production of the cytokine IL-12 in antigen-presenting cells (such as dendritic cells), thereby shifting the polarization of T cells from an (auto-)aggressive effector (Te) towards a protective or regulatory (Tr) phenotype. 1,25(OH)2D also directly exerts its immunomodulatory effects at the level of T cells.
The acquired immune system reacts in opposite ways to the native immune system (see Fig. 58-6B). Indeed, 1,25(OH)2D inhibits dendritic cell maturation and generates a coordinated action on T cell gene expression of key cytokines (IL-1, IL-2, IL-12, IL-17, INF-γ) and genes needed for antigen presentation to T cells (MHC class II and cosignaling proteins). The global effect of these immune-modulating actions is thus a down-regulation of the acquired immune system. This should have beneficial effects on the occurrence or evolution of autoimmune diseases. Vitamin D–deficient mice more easily develop a more severe type of such autoimmune diseases. For example, genetically predisposed NOD mice exposed to transient vitamin D deficiency early in life have a much higher incidence of type 1 diabetes than vitamin D–replete mice,169,178 and 1α hydroxylase–deficient mice are more prone to several types of inflammatory bowel disease.179 Moreover, 1,25(OH)2D and more potent and selective analogs can significantly reduce spontaneous or experimental autoimmune diseases in rodents, such as type 1 diabetes in NOD mice, experimental allergic encephalitis, nephritis, or inflammatory bowel disease in rodents.16,24,180
Studies in human autoimmune diseases are, however, more complicated. VDR polymorphism is not clearly related to the risk for type 1 diabetes, according to a large meta-analysis.181 However, polymorphism of the 1α-hydroxylase gene is associated with the risk of type 1 diabetes in cross-sectional as well as in family studies.182 Consistent with data from NOD mice, several epidemiologic studies in humans report that vitamin D intake in early life may reduce later risk of type 1 diabetes. Risk reduction varied between 26% with cod liver oil to 78% with 2000 IU/d, with an overall effect of 30% reduction in five published reports.180 Since 25(OH)D serum levels have not been measured in the cohorts of children, a threshold cannot be defined for optimal reduction of type 1 diabetes.
A low vitamin D status has also been repeatedly associated with a higher risk for multiple sclerosis. A large prospective, nested, case-control study among more than 7 million U.S. military personnel revealed that low 25(OH)D level was a strong risk factor for later occurrence of multiple sclerosis (odds ratio of about 2 for serum 25[OH]D <20 ng/mL, with possibly even greater “protection” by higher levels).183 However, as for type 1 diabetes, no controlled, randomized intervention studies have yet proved a causal relation between vitamin D (deficiency or insufficiency) and later occurrence of autoimmune diseases, but randomized trials (especially in genetically at-risk groups) should receive high priority. All these observations, however, suggest that preventing vitamin D deficiency in the perinatal period, early childhood, or adolescence may have long-lasting effects on autoimmune diseases. Moreover, there are several observations in mouse models that could explain such effects, such as the increased apoptosis of (autoreactive?) thymocytes or lymphocytes, the generation of regulatory T cells, and NKT cells after exposure to 1,25(OH)2D.180,184
Cardiovascular System
VDR-null mice, as well as 1α hydroxylase–null mice, develop high-renin hypertension and cardiac hypertrophy that can be prevented by treatment with an angiotensin blocker.185 In vitro or in vivo exposure to 1,25(OH)2D decreases renin production, probably by direct regulation of the gene expression via a VDRE in the promoter of renin. Observational studies in humans found an inverse association between 1,25(OH)2D levels and blood pressure or plasma renin levels in normotensive or hypertensive individuals. Prospective cohort studies reported that incident hypertension over a 4-year follow-up is lowest when the serum 25(OH)D is 30 ng/mL or higher.186 Also, in the NHANES III population, a significant negative relation between serum 25(OH)D concentrations and systolic, diastolic, and pulse pressure among the total adult population was observed.187 The results of these observational studies are supported by two randomized controlled trials in which vitamin D treatment reduced blood pressure in hypertensive subjects and elderly community-dwelling women.
VDR-null mice display increased thrombogenicity and decreased fibrinolysis when exposed to inflammatory stimuli,188 whereas 1,25(OH)2D has beneficial effects on most cells of the vascular wall. In humans, a low vitamin D status is associated with a number of cardiovascular risk factors,189 including the metabolic syndrome. The largest prospective intervention trial (WHI) with calcium (1g/d) and vitamin D (400 IU/d), however, revealed no increased nor decreased coronary or cerebrovascular risk after 7 years of follow-up.190 Several large-scale observational studies demonstrated that survival and especially cardiovascular events were lower in patients on chronic renal replacement therapy treated with 1,25(OH)2D analogs, compared with either untreated or 1,25(OH)2D-treated groups.191 However, a large meta-analysis casts doubt on this conclusion.192 In addition, vitamin D excess can have deleterious effects on all structures of the vascular wall, with ectopic calcification and organ failure of kidney, cardiac valves, myocardium, and most other soft tissues. These data suggest a beneficial effect of the vitamin endocrine system (within specific optimal limits) on cardiovascular targets but certainly need confirmation by a proper prospective, large-scale randomized trial.
Muscle and Muscle Function
VDR is expressed in myoblasts and is also present in low concentrations in mature striated muscle cells. VDR-null mice, even on a high calcium diet, show maturation problems of their muscle fibers, with smaller muscle fibers and expression of embryonic markers even after weaning.193 Genes that are typically expressed early in life (e.g., myf-5) are under negative control by 1,25(OH)2D. Evaluation of muscle performance in VDR-resistant or vitamin D–deficient mice is difficult to interpret, because hypocalcemia may have major effects on calcium fluxes in muscle cells. Patients with chronic renal failure and vitamin D deficiency (thus combined deficiency of 25[OH]D and 1,25[OH]2D) can develop severe myopathy and inability to walk that can be promptly restored by appropriate vitamin D and/or analog treatment. Sarcopenia (progressive loss of muscle mass and strength) is highly prevalent in the elderly and frequently associated with vitamin D deficiency. Vitamin D supplementation can modestly and inconsistently improve muscle function and improve body sway. Meta-analysis of several prospective intervention studies revealed that vitamin D supplementation in vitamin D–deficient elderly subjects can modestly reduce the risk of falls;194 this may explain, together with beneficial effects on bone, a reduced fracture risk.195
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


