FIGURE 87-1. Classification of thyroid nodules on the basis of results from clonality studies. Such studies imply that most thyroid nodules are “true” thyroid tumors compared with polyclonal hyperplastic nodules. Traditionally, only thyroid adenomas are considered true tumors. This is based solely on a histologic definition—the presence of a capsule and a growth pattern that is different from the surrounding normal parenchyma in an otherwise normal thyroid gland.
ENVIRONMENT VERSUS HEREDITY
Until recently, nontoxic goiter was regarded mainly as a consequence of iodine deficiency. However, a number of goitrogens6–8 and cigarette smoking are important environmental risk factors in the origin of nontoxic goiter. The impact of smoking, most likely mediated by thiocyanate, which competitively inhibits the iodide transport into the thyroid, has been extensively studied.6,9 Additional etiologically important factors consist of gender,10,11 age,12 and increased body mass index (BMI).13 The effect of a certain goitrogen is influenced by the degree of iodine sufficiency and therefore varies regionally and interindividually. However, it is most likely that interactions between environmental factors and individual genetic determinants ultimately determine the onset of the goiter.10 Nontoxic goiter appearing early in life, often clustering in families, suggests strong genetic susceptibility, whereas environmental determinants are more likely to have additive or triggering effects. However, in an individual, it may be impossible to evaluate the relative contribution of genetic predisposition and a multitude of potential environmental factors.
In contrast to sporadic goiters, caused by spontaneous recessive genomic variations, most cases of familial goiter present an autosomal dominant pattern of inheritance, indicating predominant genetic defects.14–16 Gene-gene interactions or various polygenic mechanisms (i.e., synergistic effects of several variants or polymorphisms) could increase the complexity of the pathogenesis of nontoxic goiter and offer an explanation for its genetic heterogeneity. A strong genetic predisposition is indicated by family and twin studies. Thus, children of parents with goiter have a significantly higher risk for developing goiter compared with children of nongoitrous parents.17 The high incidence in females and the higher concordance in monozygotic than in dizygotic twins also suggest a genetic predisposition.10 Moreover, preliminary evidence reveals a positive family history for thyroid disease in those who have postoperative relapse of goiter, which can occur from months to years after surgery.18,19
The development of nontoxic goiter is most likely a continuous process that starts with thyroid hyperplasia. Therefore, defects in genes that play an important role in thyroid physiology and thyroid hormone synthesis could predispose to the development of goiter, especially in cases of borderline or overt iodine deficiency. Such defects could lead to dyshormonogenesis as an immediate response, thereby indirectly explaining the nodular transformation of the thyroid as a late consequence of dyshormonogenesis, as a form of maladaptation.12 Genes that encode the proteins involved in thyroid hormone synthesis, such as the thyroglobulin gene (TG gene), the thyroid peroxidase gene (TPO gene), the sodium-iodide symporter gene (SLC5A5), the Pendred syndrome gene (SLC26A4), the TSH receptor gene (TSH-R gene), the iodotyrosine deiodinase gene (DEHAL1), and the thyroid oxidase 2 gene (THOX2), are convincing candidate genes in familial euthyroid goiter. Originally, several mutations in these genes were identified in patients with congenital hypothyroidism. However, in cases of less severe functional impairment that can still be compensated, the contribution of variants of these genes in the development of nontoxic goiter is possible. Moreover, in case of mutations in the TG gene,20,21 SLC26A4,22,23 and SLC5A5,24,25 patients with nontoxic goiter have also been identified.
LINKAGE STUDIES
To identify novel susceptibility loci, as well as to account for the coinheritance of different genomic regions, and to further improve the understanding of the genetic mechanisms that contribute to the development of nontoxic familial goiter, linkage analyses have been performed. A genome-wide linkage analysis has identified a candidate locus, MNG1 on chromosome 14q31, in a large Canadian family with 18 affected individuals.15 This locus was confirmed in a German family with recurrent euthyroid goiters.26 A dominant pattern of inheritance with high penetrance was assumed in both investigations. Moreover, a region on 14q31 between MNG1 and the TSH-R gene was identified as a potential positional candidate region26 for nontoxic goiter. However, in the earlier study by Bignell et al.,15 the TSH-R gene was clearly excluded. Furthermore, an X-linked autosomal dominant pattern and linkage to a second locus MNG2 (Xp22) was identified in an Italian pedigree with nontoxic familial goiter.27 To identify additional candidate regions, the first extended genome-wide linkage analysis was performed to detect susceptibility loci in 18 Danish, German, and Slovakian euthyroid goiter families.14 Assuming genetic heterogeneity and a dominant pattern of inheritance, four novel candidate loci on chromosomes 2q, 3p, 7q, and 8p were identified. An individual contribution was attributable to four families for the 3p locus and to one family for each of the other loci, respectively. On the basis of previously identified candidate regions and established environmental factors, nontoxic goiter can be defined as a complex disease (Fig. 87-2). However, for the first time, a more prevalent putative locus, present in 20% of the families investigated, was identified.14
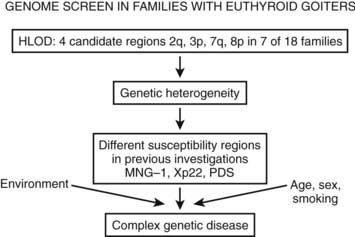
FIGURE 87-2. The identification of different susceptibility loci together with established environmental risk factors suggests that nontoxic goiter should be characterized as a complex disease. HLOD is the calculation of LOD score (logarithm of odds score) with respect to genetic heterogeneity.
The candidate region on 3p14 suggests a dominant pattern of inheritance for goiter. However, whereas linkage studies are suitable for the detection of candidate genes with a strong effect, it is possible to miss weak genetic defects of first-line candidate gene variants or of novel genes by linkage studies. Moreover, it is conceivable that the sum of several weak genetic variations in different genomic regions could lead to goiter predisposition. Therefore, the widely accepted risk factors such as iodine deficiency, smoking, old age, and female gender are likely to interact with and/or trigger the genetic susceptibility. In the future, loci identified by linkage analysis and/or association studies could reveal important genetic risk factors for familial nontoxic goiter. Further narrowing down the candidate regions and performing association studies with SNP markers in additional families and especially in case control association studies is necessary to identify the specific candidate genes for hereditary nontoxic goiter.
MUTAGENESIS AS THE CAUSE OF NODULAR TRANSFORMATION LEADING TO MULTINODULAR GOITER
Most goiters become nodular with time. From animal models of hyperplasia caused by iodine depletion,28 we have learned that besides an increase in functional activity, a tremendous increase in thyroid cell number occurs. These two events very likely orchestrate a burst of mutation events. It is known that thyroid hormone synthesis goes along with increased H2O2 production and free radical formation,29 which may damage genomic DNA and cause mutations. Together with a higher spontaneous mutation rate, a higher replication rate often will prevent mutation repair and increase the mutation load of the thyroid, thereby also randomly affecting genes crucial for thyrocyte physiology. Mutations that confer a growth advantage (e.g., TSH-R or Gsα protein mutations) very likely initiate focal growth. Hence, autonomously functioning thyroid nodules (AFTNs) are likely to develop from small cell clones that contain advantageous mutations, as shown for the TSH-R in “hot” microscopic regions of euthyroid goiters.30
Epidemiologic studies, animal models, and molecular/genetic data outline a general theory of nodular transformation. Based on the identification of somatic mutations and the predominant clonal origin of AFTNs and cold thyroid nodules (CTNs), the following sequence of events could lead to thyroid nodular transformation that occurs in three steps, as outlined in Fig. 87-3.17 First, iodine deficiency, nutritional goitrogens, or autoimmunity may cause diffuse thyroid hyperplasia. Then, at this stage of thyroid hyperplasia, increased proliferation together with possible DNA damage due to H2O2 action causes a higher mutation load (i.e., a higher number of cells bearing mutations). Some of these spontaneous mutations confer constitutive activation of the cyclic adenosine monophosphate (cAMP) cascade (e.g., TSH-R and Gsα mutations), which stimulates growth and function. Finally, in a proliferating thyroid, growth factor expression (e.g., insulin-like growth factor 1 [IGF-1], transforming growth factor β [TGF-β], or epidermal growth factor [EGF]) is increased. As a result of growth factor co-stimulation, most cells divide and form small clones. After increased growth factor expression ceases, small clones with activating mutations further proliferate if they can achieve self-stimulation. They thus can form small foci, which may develop into thyroid nodules. This mechanism may explain AFTNs by advantageous mutations that initiate growth and function of the affected thyroid cells, as well as CTNs by mutations that stimulate proliferation only (e.g., ras mutations, other mutations in the RAS/RAF/MEK/ERK/MAP cascade). Moreover, nodular transformation of thyroid tissue due to TSH-secreting pituitary adenomas31 and nodular transformation of thyroid tissue in Graves’ disease32 and in goiters of patients with acromegaly33 could follow a similar mechanism, because thyroid pathology in these patients is characterized by early thyroid hyperplasia.
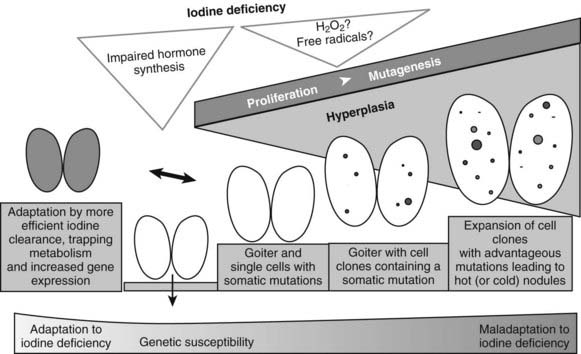
FIGURE 87-3. Hypothesis for thyroid nodular transformation. The starting point for the development of the multinodular nontoxic goiter (MNG) is hyperplasia induced by goitrogenic stimuli (e.g., iodine deficiency). Iodine deficiency increases mutagenesis directly (production of H2O2/free radicals) or indirectly (proliferation and increased number of cell divisions). Subsequently, hyperplasia forms cell clones. Some of them contain somatic mutations of the TSH-R, leading to autonomously functioning thyroid nodules (filled circles), or they contain mutations that lead to dedifferentiation and therefore cold thyroid nodules or cold adenomas (open circles).
As an alternative to the increase in cell mass, and as illustrated by those individuals who do not develop a goiter when exposed to iodine deficiency, the thyroid might also adapt to iodine deficiency without extended hyperplasia.34 Although the mechanism that allows this adaptation is poorly understood, data from a mouse model suggest an increase of mRNA expression of TSH-R, sodium iodine symporter (NIS), and TPO in response to iodine deficiency, which might be a sign of increased iodine turnover in the thyroid cell in iodine deficiency.35 Moreover, expansion of the thyroid microvasculature, caused by upregulation of vascular endothelial growth factor and other proangiogenic factors, is an additional mechanism that might help the thyroid to adapt to iodine deficiency.36
OXIDATIVE STRESS AS THE DOWNSIDE OF THYROID HORMONE SYNTHESIS
Because of the slow proliferation rate of thyroid epithelial cells, a long period (tens of years) between initiation of the tumor and appearance of MNG would seem likely. Therefore, the prevalence of thyroid tumors is a paradox that can be explained only by a high frequency of tumor initiation and/or enhanced thyroid epithelial cell proliferation. The origin of tumor formation in the thyroid therefore could be a natural or induced high mutation rate and aberrant growth stimulation in the thyroid gland. The latter very likely involves endogenous growth factors and/or exogenous goitrogenic substances.
Proliferation is very important for the manifestation of mutagenesis, and DNA replication during cell division leads to mispairing of damaged nucleotides, causing fixation of spontaneous mutations into the genome and a certain mutation load of dividing cells. Hence, compared with highly proliferating and therefore tumor-prone tissues—such as the colon, endometrium, skin, prostate, or breast—proliferation of the thyroid is rather low. If mutations occurred in the thyroid at a similar rate as in other tissues, the tumor incidence ought to be much lower than it actually is.
The main function of the thyroid gland is to synthesize the thyroid hormones L-3,5,3′,5′-tetraiodothyronine (T4) and L-3,5,3′-triiodothyronine (T3). To do so, the thyroid gland takes up iodine from food and incorporates it into thyroglobulin (Tg)—the precursor of the thyroid hormones. Iodination of tyrosyl residues on Tg requires high concentrations of H2O2 and oxidized iodine, which are generated by the enzymes thyroid oxidase 1 (THOX1), thyroid oxidase 2 (THOX2), and TPO.
Besides being a substrate in the hormone synthesis, H2O2 could be a major source of free radicals and reactive oxygen species (ROS). Because these molecules can cause substantial damage to a cell and impair normal function, thyroid epithelial cells are likely to have a potent defense mechanism to counterbalance potential damage mediated by free radicals. It has been shown that antioxidant enzymes, such as glutathione peroxidases (GPXs) or TPO, are upregulated during thyroid hormone synthesis.37 GPX3 has been suggested to directly interfere with thyroid hormone synthesis by affecting the concentration of H2O2.38 If antioxidant defense is not effective enough, excessive damage (e.g., peroxidation) should be detectable in lipids, DNA, and proteins of thyroid epithelial cells.
In thyrocytes, H2O2-mediated cytotoxicity appears to be dose dependent, requiring only low concentrations to result in thyroid cell apoptosis rather than necrosis, which could function as a barrier for tumorigenesis.39 Furthermore, findings in the thyroid glands of male Wistar rats suggest that the predominant cytotoxic response to oxidative stress might differ depending on the functional state of the gland.39 Moreover, ample evidence for excessive oxidative DNA damage has been found in the thyroid gland.40 Does this affect the spontaneous mutation rate (SMR) in the thyroid at large? It is most interesting to note that a strikingly high SMR was found in the thyroid gland of mice41: with an 8- to 10-fold higher number compared with liver, the thyroid stands out from many other tissues. Indeed, the SMR in mouse thyroid glands without any experimental mutagenic challenge shows values that usually are found only in other organs (e.g., liver) of animals treated with mutagens like ethyl nitrosourea or benzo[a]pyrene.42 The above data point to a connection between thyroid hormone metabolism, oxidative DNA damage, and SMR in the normal thyroid gland that may represent the basis for frequent tumorigenesis.
On top of this generally high mutation rate in the normal thyroid gland, environmental and lifestyle factors may add to the pool of DNA damage and increase mutagenesis and tumor initiation. Tobacco smoke, as previously mentioned,9 because it is one of the prime suspects in thiocyanate-induced blocking of iodine transport into the thyrocyte, could lead to intracellular iodine deficiency.
As outlined in the aforementioned, H2O2-mediated ROS generation is very likely to be an important starting point for thyroid tumor development (Fig. 87-4). Because iodine and H2O2 act as co-substrates in thyroid hormone synthesis, changes in iodine concentration are very likely to affect the H2O2 concentration. In fact, generation of H2O2 is inhibited by iodide in vivo and in vitro.43 H2O2 generation—which is mandatory for the organification of iodine—is, moreover, stimulated by TSH (in contrast to many other aspects of thyroid hormone synthesis, it is unclarified whether cAMP is the second messenger in H2O2 generation), which increases the expression of genes important for thyroid hormone synthesis (e.g., NIS, TPO).29 Low iodine levels and markers of increased thyroid functionality suggest activation of H2O2 generation, which could result in DNA damage and somatic mutations.44 Consequently, low iodine and high H2O2 levels should activate antioxidative defenses, which should be detectable in the cellular regulation of enzymes involved in the defense against oxidative stress.
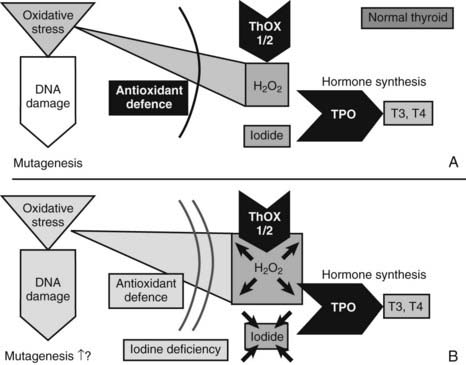
FIGURE 87-4. Mechanisms that might cause mutagenesis in the thyroid gland. The figure shows the key molecules involved in those parts of thyroid hormone synthesis which—in conditions of iodine and most likely also selenium deficiency—lead to oxidative stress, DNA damage, and possibly mutagenesis. ROS, Reactive oxygen species; THOX, thyroid oxidase; TPO, thyroid peroxidase. A, In the normal thyroid gland, the enzymes THOX1 and THOX2 generate H2O2, and TPO transfers oxidized iodine to tyrosyl residues of thyroglobulin, the precursor for T3 and T4 synthesis. H2O2 is, however, a source of ROS, which—with, other oxidative stress—can cause DNA damage. Normally, antioxidant defense could prevent oxidative stress and DNA damage. Selenoproteins like glutathione peroxidase 3 are part of the defense. B, Conditions of iodine deficiency increase levels of H2O2 and might increase the amount of oxidative stress and DNA damage. Additional selenium deficiency decreases selenoproteins and thereby could weaken antioxidative defense, which exacerbates oxidative stress and DNA damage.
Indeed, a higher expression of mRNA for superoxide dismutase 3 (SOD3)—the extracellular SOD isoform that preferentially acts in the lumen, where H2O2 is generated—is detected during experimental iodine deficiency in mice.35 Moreover, oxidative stress and antioxidant defenses are enhanced in hyperplastic and involuting glands.45
Differential expression of additional antioxidant enzymes40 underlines the importance of the antioxidant defense in the iodine-deficient thyroid gland. Moreover, glutathione peroxidases are selenium proteins. It therefore is very likely that selenium deficiency could impair the antioxidant defense and exacerbate oxidative stress (see Fig. 87-4). An increased oxidative burden in the thyroid gland through iodine deficiency is also suggested by results of the comet assay with repair-enzyme protocols to detect oxidative DNA damage.35 As a consequence, early molecular conditions for nodular and tumor transformation in the thyroid gland consist of a sequence of molecular events that include oxidative stress and DNA damage as the triggers for somatic mutations. The oxidative burden is already detectable in the normal thyroid gland and is very likely to be linked to hormone synthesis and H2O2 production. Additionally, environmental conditions (e.g., iodine deficiency) have the potential to aggravate this situation. In general, any external factor (e.g., smoking) that increases oxidative stress, causes DNA damage, or increases the SMR might aggravate the risk for tumor genesis. Also, any factor that increases proliferation (e.g., goitrogens) in all likelihood shortens the time to development of a detectable thyroid tumor.
HOT THYROID NODULES
Somatic point mutations that constitutively activate the TSH-R were first identified by Parma and coworkers in hyperfunctioning thyroid adenomas.46 However, in different studies, the prevalence of TSH-R and Gsα mutations in autonomously functioning thyroid nodules has been reported to vary from 8% to 82% and from 8% to 75%, respectively.46–49 Available studies differ in the extent of mutation detection and in screening methods. A comparison with respect to the obvious differences between studies has been done elsewhere.50,51 A comprehensive study using the more sensitive denaturing gradient gel electrophoresis52,53 revealed a frequency of 57% TSH-R mutations and 3% Gsα mutations in 75 consecutive, autonomously functioning thyroid nodules.54 These results raise the question of the molecular origin of TSH-R and Gsα mutation-negative nodules. A possible answer is given by clonal analysis of these AFTNs, which demonstrates a predominant clonal origin of thyroid nodules and implies a neoplastic process driven by genetic alteration.
In addition to the intracellular signaling network that is connected to the TSH-R, the extracellular action of different growth factors enhances the complexity of the signal flux into the thyroid cell. Growth factors like IGF-1, EGF, TGF-β, and fibroblast growth factor (FGF) stimulate growth and dedifferentiation of thyroid epithelial cells.55 Studies focused on insulin and IGF show a permissive effect of insulin and IGF-1 on TSH signaling56,57 as well as a cooperative interaction of TSH and insulin/IGF-1.58 Other studies suggest inactivation of TGF-β signaling in AFTNs due to constitutively activated TSH-R (e.g., resulting from TSH-R mutations).59 This assumption is supported by the finding of decreased expression of TGF-β1 mRNA after TSH stimulation of thyrocytes.60 Because TGF-β1 has been shown to inhibit iodine uptake, iodine organification, and thyroglobulin expression,61 as well as cell proliferation in different cell culture systems,62,63 these findings suggest that inactivation of TGF-β signaling is a major prerequisite for increased proliferation in AFTNs.64 Signal modulation of the TSH-R that would define the cause of AFTNs and the clinical phenotype therefore could take part at a number of stages and very likely involve genetic/epigenetic, gender-related, and environmental factors.
COLD THYROID NODULES
The term “cold” indicates reduced uptake on scintiscan. Because a histologic diagnosis typically is employed to exclude thyroid malignancy, many investigations of thyroid nodules refer only to the histologic diagnosis of thyroid adenoma. This histologic entity should not be confounded with the scintigraphically characterized entity “cold nodule,” which, like AFTNs or “warm nodules,” can appear histologically as thyroid adenomas or adenomatous nodules according to the WHO classification.1 In contrast, focal hyperplasia is not very well explained on the molecular level and has been discussed in detail elsewhere as the cause of thyroid tumors.65,66 A monoclonal origin has been detected for most cold thyroid nodules, which implies nodular development from a single mutated thyroid cell.67
With reference to their functional status (i.e., reduced iodine uptake), failure in the iodide transport system and failure of the organic binding of iodide were detected as functional aberrations of cold thyroid nodules long before the molecular components of iodine metabolism were known. Subsequently, decreased expression of the Na+/I− symporter (NIS) in thyroid carcinomas and benign cold thyroid nodules were suggested as the molecular mechanisms underlying the failure of iodide transport (reviewed in references 68 and 69). However, a defective cell membrane targeting the NIS protein is a more likely molecular mechanism accounting for the failure of iodine uptake in CTNs.68,70 The ultimate cause of this defect is currently unknown.
Compared with iodine transport, the organic binding of iodine is a multistep process with a number of protein components that still awaits final characterization.71 mRNA expression of enzymatic components (e.g., TPO, flavoproteins) and the substrate of iodination (i.e., Tg) have been quantified in CTNs without significant differences compared with normal follicular tissue.72,73 TPO, Tg, and thyroid-specific oxidases (THOX) have been successfully screened for molecular defects, especially in congenital hypothyroidism.74
Although CTNs could be regarded as a form of focal hypothyroidism, somatic mutations in enzymes that catalyze organic binding of iodine would need to exert a growth advantage on the affected cell to cause the development of a thyroid nodule. At least in the case of inactivating mutations in the TPO or THOX genes, growth advantage could result from a lack of enzyme activity, which would reduce not only thyroid hormone synthesis but also follicular iodide trapping in organic iodo compounds. Because these compounds have been shown to inhibit thyroid epithelial cell proliferation,75 reduced synthesis could have a proliferative effect. Therefore, somatic TPO or THOX mutations could be a molecular cause of CTN. However, mutations in the TPO gene have not been detected.76 A study of 40 cold thyroid adenomas and adenomatous nodules detected ras mutations in only a single case.67 Moreover, in the same set of CTNs, no point mutations in the mutational hot spots of the BRAF gene were detected.77 This is in line with the lack of BRAF mutations in benign follicular adenomas in other studies.78,79 So far, only one study has detected a single BRAF mutation in a set of 51 follicular adenomas.80 Moreover, the gene expression for approximately 10,000 full-length genes was compared between CTNs and their corresponding normal surrounding tissue.81 Increased expression of histone mRNAs and of cell cycle–associated genes like cyclin D1, cyclin H/cyclin-dependent kinase (CDK) 7, and cyclin B most likely reflects a molecular setup for increased proliferation in CTNs.82 In accordance with the low prevalence of ras mutations in CTNs,67 reduced expression of ras-MAPK (mitogen-activated protein kinase) cascade–associated genes was found, which might suggest minor importance of this signaling cascade. Furthermore, gene rearrangements unique to thyroid adenomas have recently been the focus (reviewed in reference 83). These studies led to the identification of the thyroid adenoma–associated gene (THADA) that encodes a death receptor–interacting protein.84 Although also reported for thyroid follicular carcinoma,85 the finding of loss of heterozygosity (LOH) at the TPO locus is characteristic for some CTNs (about 15%) but rather points to defects in a gene near TPO on the short arm of chromosome 2. Although the frequency of each of these DNA aberrations is rather low, together these chromosomal changes need to be considered in further elucidation of the molecular origin of CTNs.
Clinical Aspects
OCCURRENCE
Epidemiologic studies of multinodular goiter are hampered by problems such as selection criteria (age and gender), influence of environmental factors (e.g., iodine intake, smoking habits), evaluation of size and morphology (palpation, ultrasound, or scintigraphy), and determination of thyroid function, and whether subjects with subclinical hyperthyroidism are categorized as euthyroid or hyperthyroid. Only thyroid nodules of at least 10 mm can be identified reliably by palpation.86 With the use of ultrasound, nodules as small as 2 mm are readily detected. It therefore is not surprising that the prevalence of nodules is increased several-fold if sonographic examination is applied, because 70% of thyroid nodules disclosed by sonography are smaller than 10 mm in diameter.86,87
Most studies have focused on middle-aged women and the elderly, whereas only a few have documented the prevalence of multinodular thyroid disease in a cross-sectional investigation of the adult population in a community. Longitudinal studies covering many years are necessary to give valid figures on incidence, etiologic risk factors, and the natural history. Such studies that take the above-mentioned problems into consideration are not available. These limitations therefore should be borne in mind when the available data are considered.87 Iodine deficiency is still the most frequent single cause of multinodular endemic goiter worldwide (see Chapter 87). Considerable regional variation exists even in nonendemic goiter areas. In the Whickham survey, 16% of the cohort had simple goiter.88 In men, the prevalence declined with age from 7% in those younger than 25 years to 4% in those older than 65 years. Among women, the frequency declined from 31% in those younger than 45 years to 12% in those older than 75 years. This finding fits the observation that lean body mass, known to decline with age, is the major determinant of thyroid size.87 Illustrating the influence of iodine intake on the epidemiology of sporadic goiter, 31 of 423 (7.3%) 68-year-olds had goiter in Jutland, Denmark (low iodine intake area), versus 2 of 100 (2%) in Reykjavik, Iceland (high iodine intake area).89
A cross-sectional study of the community in Whickham found a prevalence of hyperthyroidism of 25 per 1000 women and 2 per 1000 men in an adult population.88 Others have reported similar figures.88 The yearly incidence of hyperthyroidism (all types) varies between 0.1 and 0.2 per 1000 men and between 0.3 and 1.3 per 1000 women. As with nontoxic goiter, iodine intake is of paramount importance. In Denmark, a country with a borderline sufficient iodine intake, multinodular toxic goiter accounts for 50% of patients with hyperthyroidism, whereas Iceland, with a high iodine intake, has a lower proportion of multinodular goiter (6%) and a greater number of cases of Graves’ disease.89
NATURAL HISTORY
The natural history of multinodular goiter, with respect to goiter growth and function, varies and is difficult to predict in a given patient. The spontaneous growth rate in selected populations has been estimated to be up to 20% yearly90 but usually is much lower. No specific parameter exists that can predict the growth potential of multinodular goiter, which can be accurately assessed by serial yearly measurements of the size of the goiter and individual nodules by ultrasonography.91
Painful nodules are usually the result of hemorrhage into a nodule or a cyst in the goiter. The diagnosis is readily made by ultrasonographic examination and fine-needle aspiration biopsy. Such a growing painful nodule may represent thyroid malignancy and should be investigated accordingly. Multinodular goiter is not usually associated with a significantly increased risk for the development of thyroid malignancy. The risk of malignancy in thyroid nodules occurring within a multinodular goiter has not been completely clarified, but most authors find a similar frequency in uninodular and multinodular goiters.92
Patients with nontoxic multinodular goiter can become hyperthyroid or, less commonly, hypothyroid. Hyperthyroidism in such patients often develops insidiously, in contrast to that of Graves’ disease. It often begins with a prolonged period of subclinical hyperthyroidism characterized by low serum TSH and normal serum free T4 and triiodothyronine (T3) concentrations.87 This hyperthyroid state is the consequence of goiter growth and an associated increase in the mass of autonomously hormone-producing thyroid cells. Hyperthyroidism can also be the result of an increase in iodine intake from iodine-containing drugs such as disinfectants and amiodarone or from radiographic contrast agents, which, in a goiter with increased autonomous iodine metabolism, leads to the production of excessive amounts of thyroid hormone. Little is known of the incidence and the time frame for this progression from the nodular nontoxic goiter toward the nodular toxic goiter. In a large population-based cross-sectional study in an iodine-deficient area, nodular autonomy increased with age and reached 15% in elderly people.93 It appears from a few longitudinal studies87 that within 5 years, hyperthyroidism will emerge in approximately 10% of patients with a nodular goiter. In a few cases, autonomy of some of the thyroid nodules may return.
Development of hypothyroidism in a patient with multinodular nontoxic goiter is rarer. This observation is difficult to explain, but the situation is probably caused by coexisting autoimmune thyroiditis.
DIAGNOSIS
Clinical Examination
Pertinent clinical signs and symptoms are given in Table 87-1, and diagnostic aspects are summarized in Table 87-2. For most, the thyroid gland does not become palpable until the volume has doubled. A visibly diffusely enlarged goiter has often reached a volume of 30 to 40 mL. Detection of nodules depends on their size, morphology, and location within the thyroid parenchyma, the anatomy of the patient’s neck, and the training of the physician. Among patients who present with a palpable nodule, approximately half have more than one lesion by sonographic examination.86 Sonography detects approximately five times as many nodules as thyroid palpation, and twice as many when only nodules larger than 2 cm are considered.86,87 Awareness, however, may depend on localization, speed of growth, and the possible pain or discomfort related to hemorrhage into a nodule (see Table 87-2).
Table 87-1. Clinical Signs and Symptoms of Multinodular Goiter
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
