FIGURE 114-1. Nocturnal serum concentration profiles of growth hormone (GH) illustrate the differences between the acute phase and the chronic phase of critical illness in an intensive care setting.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
Distinct Alterations Within the Somatotropic Axis During Chronic Critical Illness
In chronic critical illness, the changes observed within the somatotropic axis are different. First, the pattern of GH secretion is very chaotic, and the amount of GH, which is released in pulses, is now much reduced compared with the acute phase6,23–25 (see Fig. 114-1). Moreover, although the nonpulsatile fraction is still somewhat elevated, and the number of pulses is still high, the mean nocturnal GH serum concentrations are scarcely elevated if at all,23 compared with the healthy, nonstressed condition, and substantially lower than in the acute phase of stress.5 We observed that when intensive care patients are studied from 7 to 10 days of illness onward, in the absence of drugs known to exert profound effects on GH secretion such as dopamine,26,27 calcium entry blockers, and glucocorticoids, mean nocturnal GH levels are uniformly approximately 1 µg/L,23 trough levels are easily detectable (and thus still elevated), and peak GH levels hardly ever exceed 2 µg/L.6,23–25 These results are surprisingly independent of the patient’s age, gender, body composition, and type of underlying disease.3,5 Second, only the pulsatile fraction of GH secretion, which is substantially reduced, correlates positively with circulating levels of IGF-1, IGFBP-3, and ALS, all of which are low.6,24,25 Thus the smaller the GH pulses become, the lower the circulating levels of GH-dependent IGF-1 and ternary complex binding proteins. This clearly no longer represents a pure state of GH resistance. Serum levels of GHBP,3 assumed to reflect GH receptor expression in peripheral tissues, which are increased in patients who are critically ill for several weeks compared with those measured in a matched control group, are in line with recovery of GH responsiveness with time during severe illness.3,6 Moreover, low serum levels of GH-dependent IGF-1 and IGF-binding proteins (IGFBP-3, ALS, and IGFBP-5) are closely related to biochemical markers of impaired anabolism, such as low serum osteocalcin and leptin concentrations, during prolonged critical illness.6 These findings suggest that relative GH deficiency, epitomized by reduced pulsatile GH secretion, participates in the pathogenesis of the wasting syndrome, especially in the chronic phase of critical illness. Furthermore, there is a gender dissociation in that men show a greater loss of pulsatility and regularity within the GH secretory pattern than women (despite indistinguishable total GH output) and concomitantly have lower circulating IGF-1 and ALS levels3 (Fig. 114-2). It remains unknown whether the (paradoxical) sexual dimorphism within the GH/IGF-1 axis and the fact that males seem to be at higher risk of an adverse outcome from chronic critical illness than females3 is a casual or causal association.
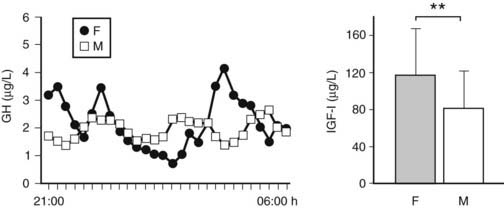
FIGURE 114-2. The more feminized pattern of growth hormone (GH) secretion (more irregular and less pulsatile GH secretory pattern for an identical mean nocturnal GH level) in prolonged critically ill men compared with women is illustrated by the representative nocturnal (21:00-06:00 hr) GH serum concentration series (sampling every 20 minutes) obtained in a male and a matched female patient. Concomitantly, prolonged critically ill men have lower circulating levels of insulin-like growth factor 1 (IGF-1) than female patients do. IGF-1 results are presented as mean ± standard deviation. **P < 0.01.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
Pathophysiology of Chronic Changes Within the Somatotropic Axis
The pathogenesis of the secretory pattern of GH in prolonged critical illness is probably complex. One of the possibilities is that the pituitary is taking part in the multiple organ failure syndrome, becoming unable to synthesize and secrete GH. An alternative explanation could be that the lack of pulsatile GH secretion is due to increased somatostatin tone and/or reduced stimulation by endogenous releasing factors such as GHRH and/or ghrelin. Studying GH responses to administration of GH secretagogues (GHRH and GHRP), in a saturating dose, enables the differentiation between a primarily pituitary origin and a hypothalamic origin of the relatively impaired GH release in prolonged critically ill patients. Indeed, the combined administration of GHRH and GHRP appears to be a most powerful stimulus for pituitary GH release in humans.28 A low GH response in critical illness would thus corroborate a pituitary dysfunction and/or a high somatostatin tone, whereas a high GH response would be compatible with reduced (hypothalamic) stimulation of the somatotropes.
GH responses to a bolus injection of GHRP have been found to be high in prolonged critically ill patients and severalfold higher than the response to GHRH, the latter being normal or often subnormal.29 GHRH + GHRP evokes a clear synergistic response in this condition, revealing the highest GH responses ever reported in a human study.29 The high GH responses to secretagogues exclude the possibility that the blunted GH secretion during protracted critical illness is due to either the lack of pituitary capacity to synthesize GH or accentuated somatostatin-induced suppression of GH release. Inferentially, one of the mechanisms that could be involved is reduced availability of ghrelin. Ultimately, the combination of low availability of somatostatin and an endogenous GHRP-like ligand such as ghrelin emerges as a plausible mechanism that clarifies (1) the reduced GH burst amplitude, (2) the increased frequency of spontaneous GH secretory bursts, (3) the elevated interpulse levels, and (4) the striking responsiveness to GHRP alone or in combination with GHRH, and this without markedly increased responsiveness to GHRH alone. Female patients with prolonged critical illness have a markedly higher response to a bolus of GHRP compared with male patients, a difference that is nullified when GHRH is injected together with GHRP3 (Fig. 114-3). Less endogenous GHRH action in prolonged critically ill men, possibly due to the concomitant profound hypoandrogenism,3 accompanying loss of action of an endogenous GHRP-like ligand with prolonged stress in both genders, may explain this finding.
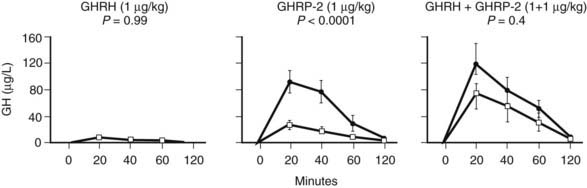
FIGURE 114-3. Responses (increments above baseline) of growth hormone (GH) obtained 20, 40, 60, and 120 minutes after intravenous bolus administration of GH-releasing hormone (GHRH) (1 µg/kg), GH-releasing peptide 2 (GHRP-2) (1 µg/kg), and GHRH + GHRP-2 (1 + 1 µg/kg) in matched male and female prolonged critically ill patients. Five men and five women were randomly allocated to each secretagogue group. Results are presented as mean ± standard error of the mean. Filled circles depict results from female patients and open squares depict results from male patients. P values were obtained using repeated measures analysis of variance.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
Effects of GH-Releasing Factors in the Chronic Phase of Critical Illness
The hypothesis of reduced endogenous stimulation of GH secretion in prolonged critical illness was further explored by examining the effects of continuous infusion of GHRP ± GHRH. Continuously infusing GHRP (1 µg/kg/h) and, even more so, GHRH + GHRP (1 + 1 µg/kg/h), for as long as 2 days was found to substantially amplify pulsatile GH secretion (more than sixfold and more than 10-fold, respectively) in this condition, without altering the relatively high burst frequency24,25 (Fig. 114-4). Reactivating pulsatile GH secretion evoked a proportionate increase in serum IGF-1 (66% and 106%), IGFBP-3 (50% and 56%), and ALS (65% and 97%), indicating peripheral GH responsiveness24,25 (see Fig. 114-4). The presence of considerable responsiveness to reactivated pulsatile GH secretion in these patients and the high serum levels of GHBP clearly delineate the distinct pathophysiologic paradigm present in the chronic phase of critical illness as opposed to the acute phase, which is thought to be primarily a condition of GH resistance. After 2 days’ treatment with GHRP, (near) normal levels of IGF-1, IGFBP-3, IGFBP-5, and ALS are reached, and as shown in a subsequent study, this normalization is maintained for at least 5 days6 (Fig. 114-5). Concomitantly, GH secretion after 5 days of treatment with GH secretagogues was found to be lower than after 2 days of treatment, suggesting active feedback inhibition loops, which most likely prevented overtreatment.6,25 In this study, in which GHRP was infused together with thyrotropin-releasing hormone (TRH) for 5 days, the self-limiting endocrine responses induced anabolism at the level of several peripheral tissues, as indicated by an increase in the serum levels of osteocalcin, insulin, and leptin and a decrease in urea production.6 Usually, infusion of GHRP without GHRH suffices to reactivate pulsatile GH secretion and to elicit the IGF-1 and IGFBP responses in prolonged critical illness. However, in critically ill men, in particular those with a very long intensive care stay, it may be necessary to add a low dose of GHRH (0.1 µg/kg/h suffices) (Van den Berghe et al., unpublished observations) because of the simultaneous lack of endogenous GHRH activity accompanying the reduced availability of the GHRP-like ligand.3
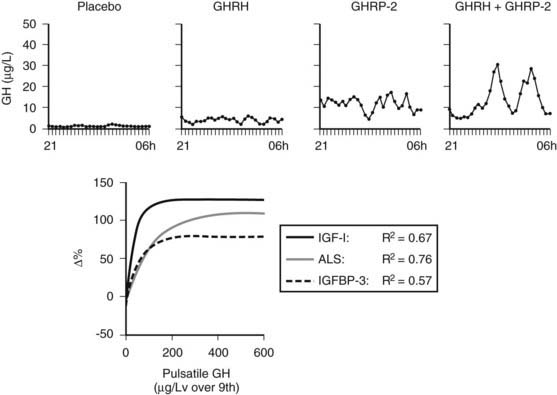
FIGURE 114-4. Nocturnal serum growth hormone (GH) profiles in the prolonged phase of illness illustrate the effects of continuous infusion of placebo, GH-releasing hormone (GHRH) (1 µg/kg/h), GH-releasing peptide 2 (GHRP-2) (1 µg/kg/h), or GHRH + GHRP-2 (1 + 1 µg/kg/h). Exponential regression lines have been reported between pulsatile GH secretion and the changes in circulating insulin-like growth factor 1 (IGF-1), acid-labile subunit (ALS), and IGF-binding protein 3 (IGFBP-3) obtained with 45-hour infusion of placebo, GHRP-2, or GHRH + GHRP-2. They indicate that the parameters of GH responsiveness increase in proportion to GH secretion up to a certain point, beyond which further increase of GH secretion has apparently little or no additional effect. It is noteworthy that the latter point corresponds to a pulsatile GH secretion of approximately 200 µg/L over 9 hours or less, a value that can usually be evoked by the infusion of GHRP-2 alone. In chronic critical illness, GH sensitivity is clearly present, in contrast to the acute phase of illness, which is thought to be primarily a condition of GH resistance.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
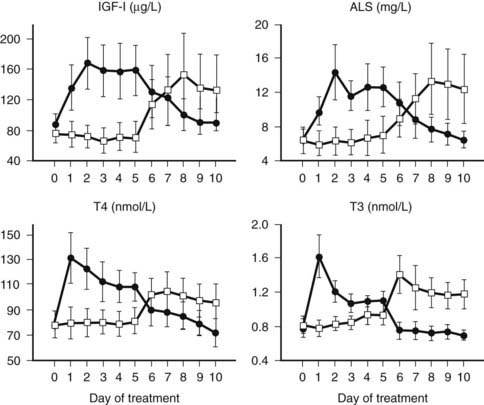
FIGURE 114-5. Serum concentrations (mean ± standard error of the mean) of insulin-like growth factor 1 (IGF-1), acid-labile subunit (ALS), thyroxine (T4), and triiodothyronine (T3) in response to a randomized treatment with either 5 days GHRP-2 + thyrotropin-releasing hormone (TRH) infusion (1 + 1 µg/kg/h) followed by 5 days of placebo (filled circles) or 5 days of placebo followed by 5 days GHRP-2 + TRH infusion (1 + 1 µg/kg/h) (open squares) in a group of 10 male and 4 female critically ill patients on ventilation in the intensive care unit. All P < 0.0001 with analysis of variance. The mean age of the patients was 68 years. The mean intensive care unit stay at the time of study start was 40 days.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
Treatment With Growth Hormone During Critical Illness
In view of the anabolic properties of GH and IGF-1, a large multicenter study investigated the effects of high-dose GH treatment in long-stay intensive care patients.30 Instead of improving outcome, this intervention doubled mortality and worsened morbidity. Although the authors of this study did not provide an explanation for the unexpected outcome, the difference between the acute and chronic stress responses may be important. The rationale for the use of high GH doses in that trial has presumably been the extrapolation, now invalidated, that all conditions of stress-associated hypercatabolism, and thus also the catabolic state of prolonged critical illness, are brought about primarily by resistance to GH in the presence of normal or adaptively altered pituitary function, and that the induction of anabolism in these conditions would thus require very high GH doses. The knowledge that is now available on the different states of the somatotropic axis in acute and prolonged critical illness clarifies, at least partially, why the administration of high GH doses to sick (but often GH-responsive) patients may have evoked side effects. Indeed, high doses of GH administered in the chronic phase of critical illness can induce IGF-1 levels into the acromegalic range, excessive fluid retention (as much as 20% of body weight), hypercalcemia, and pronounced insulin resistance with hyperglycemia.31 In view of the broad spectrum of target tissues for GH, and taking into account the preexisting impairment of vital organ functions in the critically ill, the excessive doses of GH may have further deteriorated the function of multiple organs.
A question that arises from the results of this trial is what intensive care physicians should do when patients who are GH deficient and on GH treatment become critically ill and admitted to the intensive care unit. Should GH substitution therapy be discontinued in that situation? A consensus statement from the Growth Hormone Research Society32 advises not to discontinue GH, in view of the lack of evidence that the low GH doses used for substitution therapy are harmful.
THYROTROPIC AXIS
Changes in the Acute Phase of Critical Illness
Within 2 hours after surgery or trauma, serum levels of triiodothyronine (T3) decrease, whereas thyroxine (T4) and thyroid-stimulating hormone (TSH) briefly increase (Fig. 114-6).33 Apparently, low T3 levels at that stage are mainly caused by a decreased peripheral conversion of T4 to T3.34 Subsequently, circulating TSH and T4 levels often return to “normal,” whereas T3 levels remain low. Although mean serum TSH concentrations are indistinguishable from normal at that time point, the normal nocturnal TSH surge is absent.35,36 The magnitude of the T3 decrease within 24 hours has been found to reflect the severity of illness.37,38 The cytokines TNF-α, IL-1, and IL-6 have been investigated as putative mediators of acute low T3 syndrome. Although these cytokines are capable of mimicking the acute stress-induced alterations in thyroid status, cytokine antagonism in a human model failed to restore normal thyroid function.39 Low concentrations of binding proteins and inhibition of hormone binding, transport, and metabolism by elevated levels of free fatty acids and bilirubin have been proposed as factors contributing to the low T3 syndrome at the tissue level.40 Teleologically, the acute changes in the thyroid axis may reflect an attempt to reduce energy expenditure, as happens during starvation,41 and thus as an appropriate response that does not warrant intervention. This, however, remains a controversial issue because valid data to support or refute this statement are lacking.42 Indeed, although short-term intravenous administration of T3 to patients after cross-clamp removal during elective coronary bypass grafting has been shown to improve postoperative cardiac function,43,44 the doses of T3 resulted in supranormal serum T3 levels and support but do not prove an adaptive nature of the “acute” low-T3 syndrome.
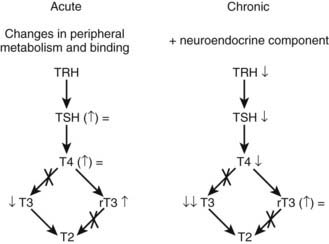
FIGURE 114-6. Simplified overview of the major changes occurring within the thyroid axis during the acute and chronic phases of critical illness. rT3, Reverse triiodothyronine; T3, triiodothyronine; T4, thyroxine; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
(From Van den Berghe G: Novel insights into the neuroendocrinology of critical illness, Eur J Endocrinol 143:1–13, 2000.)
Changes in Prolonged Critical Illness
Patients treated in intensive care units for several weeks present with a somewhat different set of changes within the thyroid axis (see Fig. 114-6). A single sample usually reveals low or low-normal TSH values and low T4 and T3 serum concentrations.45 However, overnight repeated sampling revealed that essentially the pulsatility in the TSH secretory pattern is dramatically diminished and that, as for the GH axis, it is the loss of TSH pulse amplitude that is related to low serum levels of thyroid hormone.45 Moreover, Fliers and colleagues46 elegantly demonstrated by postmortem examination of human brain specimens that when death follows chronic severe illness, the expression of the TRH gene in hypothalamic paraventricular nuclei is reduced, whereas this is not the case after death from acute insults such as lethal trauma due to a motor vehicle accident. These researchers observed a positive correlation between TRH mRNA in the paraventricular nuclei and blood levels of TSH and T3. Together, these findings indicate that production and/or release of thyroid hormones is reduced in the chronic phase of critical illness due to reduced hypothalamic stimulation of the thyrotropes, in turn leading to reduced stimulation of the thyroid gland. In line with this concept is the increase of TSH marking onset of recovery from severe illness.47 The exact mechanisms underlying the neuroendocrine pathogenesis of the low thyroid hormone levels in prolonged critical illness are unknown. Circulating cytokine levels are usually much lower at that stage,48 so other mechanisms operational within the central nervous system are presumably involved. Endogenous dopamine and prolonged hypercortisolism may each play a role, because exogenous dopamine and glucocorticoids are known to provoke or severely aggravate hypothyroidism in critical illness.49,50
Recent data indicate that in addition to the resetting of hypothalamic control, the activity in the liver of type 1 deiodinase (D1) is suppressed and of type 3 deiodinase (D3) is increased in prolonged critically ill patients. These alterations in enzyme activity were found to determine the ratio of active to inactive thyroid hormone (T3/reverse T3), indicating that changes in thyroid hormone metabolism are contributing to the picture of low-T3 syndrome in the chronic phase of critical illness.51 Interestingly, in a rabbit model of critical illness, the down-regulation of D1 and up-regulation of D3 were reversed by the simultaneous infusion of TRH and GHRP-2.52,53
It has been shown that alternative splicing gives rise to two receptor isoforms for thyroid hormone, TR-1 and TR-2. The TR-1 isoform is a bona fide T3 receptor, whereas the TR-2 acts as a dominant negative isoform. The ratio of these splice variants could therefore have a marked influence on T3-regulated gene expression, especially in view of the changing metabolism of thyroid hormone during illness. An inverse correlation between T3/reverse T3 ratio and the TR-1/TR-2 ratio in liver biopsies of prolonged critically ill patients was recently observed.54 Furthermore, higher TR-1/TR-2 ratios were present in sicker and older patients compared with the less sick and younger ones. Hence, critically ill patients appear to adapt to the decreasing thyroid hormone levels by increasing the expression of the active form of the thyroid hormone receptor gene and thereby possibly increasing the cellular sensitivity to the hormone. This constellation does not corroborate an adaptive nature of the low-T3 syndrome in prolonged critically ill patients.
Low thyroid hormone levels in protracted critical illness correlate inversely with urea production and bone degradation, which could again reflect either an adaptive, protective mechanism against hypercatabolism or a causal relationship.6 Restoring physiologic levels of thyroid hormones by continuously infusing TRH together with GHRP-2 (see Fig. 114-5), however, was found to reduce rather than increase hypercatabolism,6 an effect that was related only to thyroid hormone changes. During TRH infusion in prolonged critical illness, the negative feedback exerted by thyroid hormones on the thyrotropes was found to be maintained, thus precluding overstimulation of the thyroid axis.23,25 This self-limitation may be extremely important during critical illness to avoid hyperthyroidism, which would inadvertently aggravate catabolism. The co-infusion of TRH and GH-releasing factors appears to be a better strategy than the infusion of TRH alone because the combination, but not TRH alone, avoids an increase in circulating reverse T3.6,23 The latter is in line with the effects that TRH and GHRP-2 exert on the activity of type 1 and 3 deiodinases53,55 and eventually with other important interactions among different anterior pituitary axes for optimal peripheral responses.56
Treatment With Thyroid Hormone or Releasing Factors During Prolonged Critical Illness
It remains controversial whether correction of the illness-associated low serum and tissue concentrations of T3 by either T4 or T3 administration is required to improve clinical problems distinctively associated with prolonged critical illness.57,58 Pioneering studies with T4 administration so far have failed to demonstrate clinical benefit within an intensive care setting, but in view of the impaired conversion of T4 to T3, this is not really surprising.59,60 A recent report on thyroid hormone treatment using substitution doses of T3 in dopamine-treated pediatric patients after correction of a congenital cardiac anomaly revealed improvement in postoperative cardiac function.61 In contrast to treatment with thyroid hormones, infusing TRH allows peripheral shifts in thyroid hormone metabolism during intercurrent events and, accordingly, permits the body to elaborate appropriate concentrations of thyroid hormones in the circulation and at the tissue level, thus setting the scene for a safer treatment than the administration of T3.25 Also, the peripheral tissue responses to the normalization of serum concentrations of IGF-1 and binding proteins as evoked by GHRP infusion seem to depend on the co-infusion of TRH and the concomitant normalization of the thyroid axis. Indeed, GHRP-2 infused alone evokes identical increments in serum concentrations of IGF-1, IGFBP-3, and ALS but is devoid of the anabolic tissue responses that are present with the combined infusion of GHRP and TRH.23 Outcome benefit of TRH infusion alone or in combination with GH secretagogues in prolonged critical illness is yet to be studied.
The diagnosis of preexisting thyroid disease and its management during critical illness also can be extremely difficult, and recommendation for clinical practice is often not evidence based. In view of the hypothalamic-pituitary suppression occurring in the chronic phase of critical illness, in patients with and without previous endocrine disease, it is virtually impossible to diagnose preexisting central hypothyroidism during intensive care. Patients with preexisting primary hypothyroidism, myxedema coma being the extreme presentation, are expected to reveal low serum levels of T4 and T3 in combination with very high TSH concentrations. However, when primary hypothyroidism and severe nonthyroidal critical illness coincide, the TSH increase may be absent. Indeed, the nonthyroidal critical illness evokes the sum of changes within the hypothalamus-pituitary-thyroid axis as described previously. A decrease in serum T3 and an increase in serum reverse T3 are the most common changes in acute nonthyroidal critical illness, but serum T3 may be undetectable and serum T4 may also be dramatically reduced in patients with protracted nonthyroidal critical illness. Therefore, in patients with myxedema coma and severe comorbidity (e.g., pneumonia, sepsis), serum T3 and T4 levels are very low but could be indistinguishable from those values observed in prolonged nonthyroidal critical illness. Whereas serum TSH is markedly increased in uncomplicated primary hypothyroidism, it is paradoxically normal or even decreased in severely ill patients. Therefore, serum TSH may be lower than anticipated, from the severe hypothyroid condition of the patient with myxedema coma and concomitant illness or even frankly low. Thus a high serum TSH concentration, when observed, is in agreement with primary hypothyroidism, but a normal or low TSH does not exclude it during intercurrent critical illness. Indeed, serum TSH may be paradoxically low in this setting because of concomitant nonthyroidal critical illness, especially in patients given high-dose corticosteroids and/or dopamine. Other iatrogenic factors causing hypothyroidism, particularly in a surgical intensive care unit, are iodine wound dressings, iodine-containing contrast agents used for radiologic imaging, and drugs such as somatostatin and amiodarone. The finding of a high ratio of T3 to T4 in serum, a low thyroid hormone–binding ratio, and a low serum reverse T3 may favor the presence of primary hypothyroidism, whereas opposite changes occur in nonthyroidal critical illness. However, the diagnostic accuracy of any of these measurements is limited, and in many patients, no definite laboratory diagnosis can be established. In these patients, history, physical examination, and the possible presence of thyroid autoantibodies may give further clues to the presence or absence of thyroid disease. Repeated thyroid function tests after improvement of the nonthyroidal illness are required to confirm the diagnosis.
When and how to treat primary hypothyroidism during the course of an intercurrent nonthyroidal critical illness also remain controversial. One exception, however, is a presumed diagnosis of myxedema coma, for which there is general agreement that patients should be treated with a parenteral form of thyroid hormone. The proper initiation of thyroid hormone replacement therapy, however, is controversial because controlled studies on the optimal treatment regimen are lacking. The first uncertainty relates to the type of thyroid hormone to be given: Should it be T4 alone, T3 alone, or a combination of both? The second uncertainty is the optimal initial dose of any thyroid hormone–replacement regimen. Many clinicians prefer a loading dose of as high as 300 to 500 µg of intravenous T4 to quickly restore circulating levels of T4 to approximately 50% of the euthyroid value,62,63 followed by 50 to 100 µg of intravenous T4 daily until oral medication can be given. Higher doses do not seem to be beneficial, although Kaptein and associates64 found no increased cardiovascular risk in severely ill hypothyroid patients treated with larger doses of T4.
Some authors have advocated the use of T3 in addition to T4 because T3 does not require conversion by 5′-deiodinase enzymes to a biologically active form. In an animal experimental study by Escobar-Morreale and colleagues,65 replacement therapy for hypothyroidism with T4 alone did not ensure euthyroidism in all tissues, and a subsequent study showed that only the combined treatment with both T4 and T3 induces euthyroidism in all tissues. Tissue-specific deiodinase activity acting as local regulatory mechanisms may explain these findings. In a more recent study in patients with hypothyroidism, it appeared that partial substitution of T3 for T4 may improve mood and neuropsychological function, possibly by the increased bioavailability of T3 in the central nervous system.66 While these fascinating results await confirmation by others, replacement therapy with a combination of T4 and T3 in compensated hypothyroidism remains an experimental modality.67
The author’s experimental protocol for thyroid hormone therapy during intensive care of presumed hypothyroidism, either preexisting or iatrogenically induced when reversal of the iatrogenic cause appears impossible, advises administering a 100- to 200-µg bolus of T4 intravenously per 24 hours combined with T3 at 0.6 µg/kg ideal body weight per 24 hours in a continuous intravenous infusion, targeting serum thyroid hormone levels in the low-normal range.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


