FIGURE 21-1. Amino acid structure of vasopressin and oxytocin. Note that only positions 3 and 8 have different amino acids.

FIGURE 21-2. Factors governing arginine vasopressin (AVP) secretion. CVO, Circumventricular organ (site of osmoreceptors); PVN, paraventricular nuclei; SON, supraoptic nuclei.
In addition to magnocellular neurons, which project from the SON and the PVN to the posterior pituitary, parvocellular vasopressinergic neurons are present in the PVN and the suprachiasmatic nucleus.2 These smaller neurons project via the median eminence to the portal vascular system of the anterior pituitary while secreting vasopressin and corticotropin-releasing factor (CRF); they stimulate secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. Some parvocellular vasopressinergic neurons also project from the PVN to the forebrain, brain stem, and spinal cord. Vasopressin is released into the central nervous system, where it acts as a neurotransmitter and/or a neuromodulator.3
The gene that encodes the AVP precursor is expressed mainly in the hypothalamus but also in other tissues, including adrenal glands, gonads, cerebellum, and pituicytes in the posterior pituitary. It is structurally closely related to the gene that codes for the oxytocin (OT) precursor, which is expressed in different populations of hypothalamic magnocellular neurons. The vasopressin and oxytocin genes are closely linked in a tail-to-tail orientation on chromosome 20 with an intergenic region of 12 kb.4 Significant posttranslational processing is needed to form the mature hormone. The posttranslational product prepro-AVP-NPII is the precursor from which AVP is derived.4 The precursor consists of a signal peptide, AVP, neurophysin II (NPII; 95 amino acids), and a glycopeptide, copeptin (39 amino acids).
Neurophysin II is cleaved from prepro-AVP-NPII but remains complexed together with AVP before it is secreted into the bloodstream by exocytosis. Vasopressinergic neurons open voltage-gated calcium channels in nerve terminal membranes. This transient calcium ion influx results in fusion of neurosecretory granules with the nerve terminal membrane, as well as release of AVP and NPII in equimolar amounts into the circulation.5 Copeptin, although of unknown biological significance, may be a stable indirect marker of AVP release.
Magnocellular neurons in the supraoptic and paraventricular nuclei synthesize vasopressin within their cell bodies. The hormone is carried within neurosecretory granules down the predominantly unmyelinated axons of the magnocellular neurons, in the supraoptic-hypophyseal tract, at a speed of 2 mm per hour. The precursor undergoes successive cleavage by basic endopeptidases.6 These axons terminate in the posterior pituitary, which is rich in fenestrated capillaries.
CELLULAR ACTION OF VASOPRESSIN
Vasopressin binds to seven transmembrane, G-protein–coupled receptors on the plasma membrane of target cells. Three subtypes of vasopressin receptor exist (V1-V3) with distinct distribution, actions, and signal transduction.7,8
The V1 Receptor
The V1 receptor is found in vascular smooth muscle, liver, platelets, and multiple sites in the central nervous system. It is a 418 amino acid protein linked to the phosphinositol signaling pathway. Binding of AVP to the receptor causes activation of Gq/11-mediated phospholipase C, resulting in an increase in intracellular calcium.
Binding of AVP to V1 receptors at physiologic plasma concentrations has been shown to exert a weak pressor effect.8a The vasocontriction seen with vasopressin is most notable in the splanchnic, renal, and hepatic artery beds9–11 and has given rise to the successful use of the hormone in the treatment of bleeding esophageal varicies. At higher plasma concentrations, for instance, in response to significant hypotension or hypovolemia, vasopressin has been shown to have a significant pressor effect.12 A highly selective V1 vasopressin analogue has been shown to have similar effects in patients with sepsis.13 Rebound hypotension on withdrawal of treatment coupled with concerns regarding reduced cardiac output, pulmonary hypertension, hyponatremia, and bowel ischemia, has limited the widespread use of vasopressin in the setting of septic shock.
The V2 Receptor
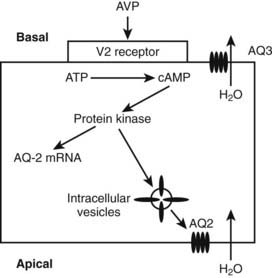
The V2 receptor is predominantly expressed on the basolateral membrane of the distal convoluting tubule and collecting ducts. Binding of AVP to renal V2 receptors stimulates the recruitment of selective water channels (aquaporins), which allow reabsorption of renal tubular water and concentration of urine. Extrarenal effects of V2 activation include factor VII release from hepatocytes and release of von Willebrand factor from vascular endothelium. AVP also causes activation of liver glycogen phosphorylase with release of glucose into the circulation.
When AVP binds to the V2 receptors on the basolateral membrane of the cells of the collecting ducts, it triggers an increase in cytoplasmic cyclic adenosine monophosphate (cAMP), which activates protein kinase (Fig. 21-3). Subsequently, movement of stored aquaporin-2 to the luminal membrane of the cell occurs. Aquaporin-2 forms tetramers in the cell membrane, with each monomer acting as a water channel14; this allows passage of tubular water into the cell. Free water reabsorption from the distal nephron and thus urine concentration are directly influenced by serum vasopressin levels. This relationship was established prior to the discovery of aquaporins. The AVP/V2 receptor interaction also simultaneously causes an increase in expression of the aquaporin-2 gene, with increased generation of mRNA for aquaporin-2.
The V3 Receptor
The V3 receptor is found on corticotropic cells in the anterior pituitary. In isolation, AVP is a weak ACTH secretagogue, but when it acts synergistically with CRF, it causes significant secretion of ACTH, which is physiologically important. Parvocellular neurons, which coexpress AVP and CRF, project via the median eminence and terminate in the hypophyseal portal bed, which provides circulation to the anterior pituitary. AVP and CRF expression by these parvocellular neurons is subject to negative feedback control by glucocorticoids.15–17 AVP has no secretagogue properties with respect to the other anterior pituitary hormones.
Regulation of Vasopressin Release
OSMOREGULATION OF VASOPRESSIN RELEASE
Plasma osmolality in healthy man varies by only 1% to 2% in physiologic conditions where there is free access to water. This precise regulation of plasma osmolality is maintained by the homeostatic process of osmoregulation.18
Changes in plasma osmolality are detected by specialized osmoreceptor cells in the anterior hypothalamus (see Fig. 21-2). Fenestrations in the blood-brain barrier cause the magnocellular cells of the circumventricular organs—the subfornical organ and the organum vasculosum lamina terminalis—to be bathed in plasma. Changes in plasma osmolality cause these hypothalamic nuclei to depolarize, sending neural signals, via the nucleus medianus, to the supraoptic and paraventricular nuclei. Elevation in plasma osmolality causes depolarization of these nuclei, leading to increased synthesis of AVP and secretion of AVP from the posterior pituitary. The osmoreceptor cells are solute specific, in that they respond vigorously to changes in plasma sodium concentration but less so to variations in blood urea.19 They respond to mannitol but are completely insensitive to changes in blood glucose concentration in experimental situations.
A linear relationship has been noted between plasma osmolality and plasma AVP concentrations throughout the physiologic range of plasma tonicity.20–22 If plasma concentrations are lowered by excessive ingestion of hypotonic fluid to below 280 to 285 mOsm/kg, secretion of AVP is suppressed, and plasma concentrations of the hormone are undetectable on radioimmunoassay measurement. In the absence of AVP-mediated water reabsorption from the kidney, hypotonic polyuria develops.22 The increase in free water clearance allows plasma osmolality to rise into the normal range. If plasma osmolality rises, owing to dehydration, plasma concentrations of AVP increase in proportion to the rise in plasma osmolality. The action of AVP in concentrating the urine and allowing reabsorption of water restores plasma osmolality to normal. This homeostatic process occurs continuously to maintain plasma osmolality within a narrow reference range. The relationship between plasma AVP concentration and urine osmolality is shown in Fig. 21-4.
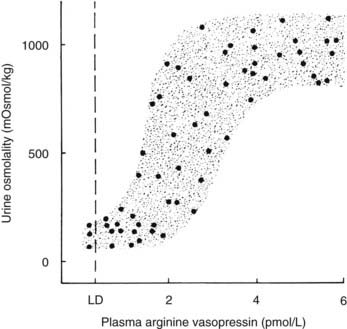
FIGURE 21-4. Relation between plasma arginine vasopressin (AVP) (pAVP) and urine osmolality. Data were obtained during water loading and fluid restriction in a group of healthy adults. Maximal urine concentration is achieved by pAVP values of 3 to 4 pmol/L. LD, Limit of detection of the AVP assay, 0.3 pmol/L.
The development of sensitive radioimmunoassays has allowed the nature of this relationship to be studied experimentally. When intravenous infusion of hypertonic sodium chloride solution is used to increase plasma osmolality in healthy volunteers, plasma AVP can be shown to increase in a linear fashion. Application of linear regression analysis to the data shows that a direct correlation defines the relationship between plasma osmolality and plasma AVP concentration (Fig. 21-5). The qualitative relationship is expressed by the following equation:
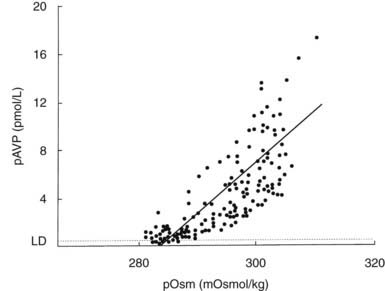
FIGURE 21-5. Relation between plasma osmolality (pOsm) and plasma arginine vasopressin (AVP) (pAVP). Increases in pAVP in response to hypertonicity induced by infusion of 855 mmol/L saline in a group of healthy adults. The mean regression line (dashed line) is defined by the following equation: pAVP = 0.43 (pOsm 284); r = 0.96; P < .001. LD, Limit of detection of the AVP assay, 0.3 pmol/L.
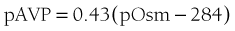
where pAVP indicates plasma AVP concentration, and pOsm indicates plasma osmolality.23
This mathematical formula describes two important physiologic characteristics of the relationship between plasma osmolality and plasma AVP that are clinically relevant. The first of these is the osmotic threshold for AVP release, which is defined by the abscissal intercept of the regression line. At a mean plasma osmolality of 284 mOsm/kg, plasma AVP is secreted, with increments in secretion as plasma osmolality rises. This represents the “set point,” or the osmotic threshold for AVP release, which is identical in man to the osmotic threshold for the onset of thirst.23 This is an important physiologic concept, as hypotonic polyuria develops when plasma osmolality is suppressed by water ingestion to concentrations below this “set point.” Therefore, AVP detectable in the plasma by radioimmunoassay at plasma osmolalities below this is, by definition, “inappropriately” elevated. Measurements of plasma AVP with ultrasensitive cytochemical methods at plasma osmolalities below the physiologic threshold have led to suggestions that secretion cannot be completely suppressed by hypotonicity,24 although the very low concentrations demonstrated experimentally could equally represent incomplete clearance of pre-secreted AVP.
The slope of the regression line, which represents the change in plasma AVP concentration per unit change in plasma osmolality, is the second important characteristic of the formula. This defines the sensitivity of the osmoreceptor-AVP releasing unit. Steeper slopes indicate a larger rise in plasma AVP concentrations after osmotic stimulation. Shallow slopes indicate lower rates of AVP secretion in response to hyperosmolality; a pathologic example of this can be seen in partial hypothalamic diabetes insipidus.25
In a few rare cases, complete disconnection of AVP-secreting neurons from their osmoreceptors has been described. The pathologic abnormality is persistent low-grade AVP release, despite plasma osmolalities that may fall below the osmotic threshold.26 AVP secretion can be increased to above this “basal” rate by stimulation of osmoreceptors, but secretion is never switched off entirely. It is hypothesized that some cells exert an inhibitory effect on AVP release, and if these cells malfunction, AVP secretion is abnormally continuous.
The osmotic threshold and sensitivity of AVP release vary significantly between individuals, but repeat testing has shown good reproducibility of these parameters within individuals.27 Studies in twins have suggested that the characteristics of the osmoregulatory line may be genetically determined, given that they are similar in monozygous but not dizygous twins.28
Although physiologic control of AVP secretion and thirst is almost entirely osmotic, the switch-off of the two is nonosmotic and is triggered by the act of drinking. In studies in healthy men who have been rendered hyperosmolar by hypertonic saline infusion29 or by dehydration,30 drinking is associated with an immediate fall in plasma AVP and thirst, before any changes in plasma osmolality can be measured. The fall in plasma AVP is so rapid that it mimics the half-life of AVP,29 suggesting that a neuroendocrine reflex, stimulated by oropharyngeal distention, switches off AVP secretion.
The conventional relationship between plasma osmolality and plasma AVP concentration is altered in a number of physiologic and pathophysiologic situations. Pregnancy causes a lowering of the threshold for AVP secretion in both rats31 and humans,32 which is responsible for the fall in basal plasma sodium concentration during pregnancy. The sensitivity of the osmoregulatory line remains unchanged. The mechanism is unclear but may be related to increased plasma concentrations of human chorionic gonadotropin (hCG). In the luteal phase of the ovulatory cycle, a small but significant decrease in plasma osmolality also occurs as a result of downward resetting of the osmotic thresholds for thirst and AVP release.33
Normal physiologic aging is associated with complex changes in osmoregulation.34 Basal circulating AVP concentrations increase with age, and the AVP response to osmotic stimulation has been shown to be enhanced in comparison with that in younger subjects.35,36 Thirst, as measured by visual analogue scales, is attenuated, however, and measured water intake after osmotic stimulation intake is reduced.37 This makes elderly humans more vulnerable to hypernatremia; many elderly patients in long-term care institutions exhibit permanent hypernatremia, although cognitive impairment may contribute to reduced thirst in these individuals.38
Osmoregulation in diabetes mellitus seems normal,39 even in the presence of hypernatremia,40 with preserved solute specificity of both AVP release and thirst; hyperglycemia stimulates neither thirst nor AVP secretion. Chronic hyperglycemia renders the renal tubules resistant to the antidiuretic effects of AVP, however, as the result of failure of recruitment of aquaporin-2.41 Improved glycemic control reverses the state of partial nephrogenic diabetes insipidus.41 Survivors of HONK, however, show typical osmoregulatory changes of aging, with exaggerated AVP secretion and attenuated thirst in response to dehydration.42
BAROREGULATION OF VASOPRESSIN RELEASE
Although small variations in plasma osmolality can have profound effects on AVP secretion, physiologic fluctuations in blood pressure have almost no effect on plasma AVP secretion. However, pathophysiologic falls in both blood volume and pressure can stimulate AVP secretion. Although small falls in arterial blood pressure (on the order of 10%) have been shown to slightly increase plasma AVP concentrations (see Fig. 21-5), larger falls of 20% to 30% are required to stimulate sufficient AVP to exert a compensatory pressor effect.43,44 This makes teleologic sense as falls in blood pressure of up to 10% are common during a daily cycle, and if AVP were stimulated by these small falls, humans would be in a permanent state of antidiuresis. The relationship between blood pressure and plasma AVP is shown in Fig. 21-6.
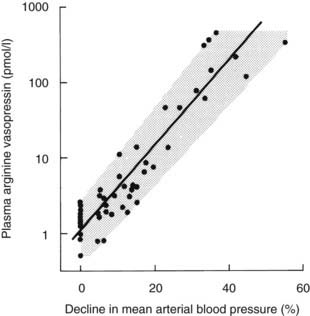
FIGURE 21-6. Relation of plasma arginine vasopressin (AVP) (pAVP) to the percentage decline in mean arterial blood pressure (MABP). Arterial blood pressure was reduced by infusing increasing doses of trimetaphan in healthy men. The regression line is defined by the following equation: log (pAVP) = 0.06 (MABP 0.67); r = 0.98; P < .001; N = 48.
The baroreceptors are situated in the cardiac atria, the carotid sinus, and the aortic arch. They exert a tonic inhibitory effect on AVP secretion in normal circumstances, but when they are unloaded during hypotension or hypovolemia, they can stimulate very high plasma concentrations of AVP via pathways that are separate from the osmoreceptors. The relation between blood pressure and AVP is exponential rather than linear and can be modified by neurohumoral influences such as atrial natriuretic peptides, which inhibit AVP responses, and norepinephrine, which augments baroregulatory AVP responses.45 Baroregulatory inputs also modify osmotic AVP secretion, in that hypovolemia augments the AVP response to hypernatremia.46,47
OTHER MECHANISMS REGULATING VASOPRESSIN RELEASE
In addition to osmotic and baroregulatory stimuli, AVP secretion occurs in response to nausea,48,49 manipulation of abdominal contents at surgery,50 and hypoglycemia.51 All of these secretagogues stimulate AVP secretion independently of the osmotic pathways.
Diabetes Insipidus
Diabetes insipidus is a clinical syndrome characterized by hypotonic polyuria due to failure to concentrate urine. Urine flow rates in excess of 40 mL/kg per 24 hours in adults, or more than 100 mL/kg per 24 hours in infants, are suggestive of diabetes insipidus. Three major categories of diabetes insipidus have been identified.52
Polyuria is caused by insufficient AVP secretion, so that there is no effective antidiuretic hormone. There needs to be a loss of at least 80% of the magnocellular neurons that synthesize and secrete AVP before polyuria develops.
In nephrogenic diabetes insipidus, AVP secretion is normal, but renal insensitivity to the antidiuretic effects of the hormone is present.
In dipsogenic diabetes insipidus, excessive fluid intake reflects abnormal perception of thirst.
A full list of causes of diabetes insipidus is shown in Table 21-1.
Table 21-1. Causes of Diabetes Insipidus
| Hypothalamic DI | |
| Congenital | |
| Acquired | |
| Nephrogenic DI | |
| Congenital | Hereditary (X linked recessive or AD) |
| Acquired | |
| Dipsogenic DI | |
AD, Autosomal dominant; DI, diabetes insipidus; DIDMOAD, diabetes insipidus, diabetes mellitus, optic atrophy, and deafness; GI, gastrointestinal; TB,tuberculosis.
HYPOTHALAMIC DIABETES INSIPIDUS
HDI (also known as neurogenic, central, or cranial DI) occurs when AVP secretion is insufficient to prevent the development of a hypotonic diuresis, such that the patient presents with polyuria and nocturia. The lesion in the vast majority of cases spares the thirst mechanism53; therefore, because thirst appreciation is intact, patients respond to the polyuria with appropriate polydipsia. The increase in fluid intake is nearly always sufficient to maintain normonatremia; hypernatremia in which fluid is freely available should always raise the suspicion of associated thirst abnormalities. The AVP deficiency may be complete, with no detectable plasma AVP, or partial, where AVP is detectable in the plasma at low concentrations inappropriate to the ambient plasma osmolality.
HDI is rare in the general population with an estimated prevalence of 1:25,000 but is common in certain subgroups, such as patients undergoing pituitary surgery, or transiently in other forms of neurosurgical intervention.
Most cases of HDI are acquired during adult life, and the most common cause of DI in most endocrine practices is surgery for pituitary tumors. Pituitary adenomas rarely cause DI, and when a pituitary mass is associated with polyuria, then craniopharyngioma, granuloma, and hypophysitis are far more likely diagnoses.
DI occurs in the immediate postoperative period in 18% to 30% of cases,54–57 with most cases presenting in the first 2 days following surgery. The most common natural history is spontaneous resolution over the next 2 to 5 days. The pathophysiology of acute transient DI is thought to be surgical contusion injury to the magnocellular neurons projecting from the supraoptic and paraventricular nuclei to the posterior pituitary. In some patients however, DI persists and AVP deficiency becomes permanent. Most neurosurgical series report a low incidence of 1% to 8% for permanent DI, following transsphenoidal surgery.54,56–58 Many published studies are retrospective, however, and use polyuria alone as the criteria for diagnosis of DI. We found that formal water deprivation testing identified permanent DI in 30% of patients who had transcranial surgery for pituitary adenoma and much higher figures of over 90% following craniopharyngioma surgery.55
In a small minority of patients, post-hypophysectomy DI follows the triple phase response. Initially, classical postoperative DI presents in the first 2 days following surgery. However, after 4 to 8 days of hypotonic polyuria, urine output drops and plasma sodium begins to fall, even after discontinuation of desmopressin therapy. This second, antidiuretic phase is characterized by a rise in plasma AVP concentrations, presumably from uncontrolled release of pre-stored peptide from damaged magnocellular neurons. If hypotonic intravenous fluids continue to be administered, the fall in plasma sodium concentration can be abrupt. The antidiuretic phase of the triple phase response usually lasts 5 to 7 days, but considerable variation is noted, from 2 to 14 days, before progression to the final phase, the development of permanent DI. It is thought that permanent DI occurs when gliosis of damaged neurons prevents further AVP release.
The major factor influencing the risk of developing diabetes insipidus following pituitary surgery is tumor type. Craniopharyngioma is associated with preoperative DI, but in addition, surgical intervention is far more commonly associated with the development of DI than is surgery for any other intracranial tumor.55 Surgery for ACTH-secreting pituitary adenomas has been associated with a higher risk of developing DI in a number of series.54,56,58 Data from the literature are conflicting regarding the influence of tumor size on the risk of postoperative diabetes insipidus. Although pituitary adenomas rarely cause HDI, metastatic deposits in the hypothalamus, or more often the pituitary stalk, can result in HDI. Metastatic deposits usually have no endocrine effects, even when they are found in the stalk or the pituitary itself, but some, particularly from carcinoma of the breast or bronchus, produce HDI. Other tumors such as germinoma, pinealoma, and parasellar meningioma can cause HDI. It is interesting to note that although radiotherapy for nonpituitary cranial tumors commonly causes anterior pituitary dysfunction, it does not seem to cause DI.59
Lymphocytic infiltration of the neurohypophysis (infundibulo-neurohypophysitis), recognized by a thickened pituitary stalk and inflammatory infiltrates of T lymphocytes and plasma cells with eosinophils, is a well-described cause of HDI.60 HDI is present in up to 30% of adult patients with Langerhans’ cell histiocytosis (LCH).61
Idiopathic DI is diagnosed less often, as better imaging techniques have been introduced and awareness of autoimmune causes has increased.62,63 It has been estimated that one third of patients previously diagnosed to have idiopathic HDI may an autoimmune origin of the condition. The characteristics of autoimmune DI include the presence of circulating antibodies against AVP-secreting cells, young age of onset, and thickened pituitary stalk T1-weighted magnetic resonance imaging (MRI).64 Patients with autoimmune DI have an increased incidence of other organ-specific autoimmune endocrine disease, particularly thyroid disease.
Traumatic brain injury causes acute diabetes insipidus in 20% of cases,65 some of which result in a triple phase response similar to that seen after pituitary surgery. DI following brain injury is nearly always transient,66 however, and only 7% of long-term survivors of head injury have permanent DI when formally tested with water deprivation.67
Sheehan’s syndrome is now rare and is not usually associated with HDI, even when anterior hypopituitarism is widespread, but maximal urine-concentrating ability is impaired in some patients.68 HDI can occasionally occur in normal pregnancy as the result of increased activity of circulating vasopressinase, the placental aminopeptidase.69 Symptoms resolve post partum. The actions of vasopressinase can unmask partial HDI in patients with pituitary disease or can worsen symptoms in undiagnosed partial HDI. HDI in pregnancy must be differentiated from the transient NDI that is seen very occasionally.70
Familial HDI is extremely rare. Autosomal dominant HDI (adHDI) is associated with loss-of-function mutations in the AVP gene, resulting in the production of a folding-incompetent peptide precursor that accumulates in the secretory pathway apparatus of magnocellular neurons.71,72 This mutant precursor is responsible for an autophagocytic process that causes progressive damage and loss of VP neurons. A single mutant allele is sufficient for this process to occur. Most mutations affect exons 1 and 2 of the AVP gene.73–75 Familial HDI almost always presents in childhood.76
The Wolfram, or DIDMOAD, syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness) is a rare autosomal recessive, progressive neurodegenerative disorder characterized by the association of HDI with diabetes mellitus, optic atrophy, and bilateral sensorineural deafness, although other manifestations may include gonadal failure, renal outflow tract dilatation (secondary to reduced nerve fibers in the bladder wall), and progressive ataxia with brain stem atrophy.77 The syndrome is associated with premature death, usually due to ascending renal tract infection secondary to atonic bladder and hydronephrosis. Although DI is often reported to occur in only one third of cases, in one series where careful testing with AVP responses to hypertonic saline was performed, subnormal responses were seen in all patients.78 WS is associated with loss-of-function mutations in the WFSI gene on Ch.4p16.1, which encodes an 890 amino acid glycoprotein (wolframin) found in the endoplasmic reticulum. An additional locus for WS has been identified recently at Ch.4q22-24, suggesting that the syndrome may be genetically heterogeneous.79
NEPHROGENIC DIABETES INSIPIDUS
Nephrogenic diabetes insipidus (NDI) occurs when renal resistance to the antidiuretic effects of VP allows hypotonic diuresis to develop. An outline of potential causes is given in Table 21-1. Lithium therapy is the most common cause of NDI in clinical practice, occurring in up to 30% of patients. Although polyuria usually disappears with cessation of treatment, a minority of patients develop interstitial nephritis, and permanent NDI occurs. Hypokalemia and hypercalcemia can produce acquired NDI, which is reversible with correction of the metabolic abnormality. Poorly controlled diabetes mellitus causes renal resistance to AVP, which seems to be due to failure to recruit aquaporin-2. The reversibility of the metabolic causes of NDI shows how the generation of aquaporins can be vulnerable to metabolic and pharmacologic insults.
Familial NDI is rare. X-linked recessive familial NDI (X-FNDI) is caused by inherited loss-of-function mutations in the V2-R. More than 70 different mutations have been described, affecting all aspects of receptor physiology, including expression, ligand binding, and G protein coupling. A vast majority cause complete loss of function and present in infancy, although some mutations are associated with partial loss of function.80 A minority of FNDI families have autosomal recessive (AR-FNDI), loss-of-function mutations of the gene encoding AQP2. Most mutations occur in the region of the gene that encodes the transmembrane domain of the water channel protein. Other kindreds have been described with an autosomal dominant NDI (AD-NDI) caused by loss-of-function mutations in AQP2 affecting the carboxyl-terminal intracellular domain of the protein. NDI is expressed in these kindreds because the protein product of the mutant allele forms mixed oligomers with the product of the wild-type allele, resulting in sequestration within the Golgi or mistargeting to the basolateral rather than the apical membrane in a dominant negative manner.81,82
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


