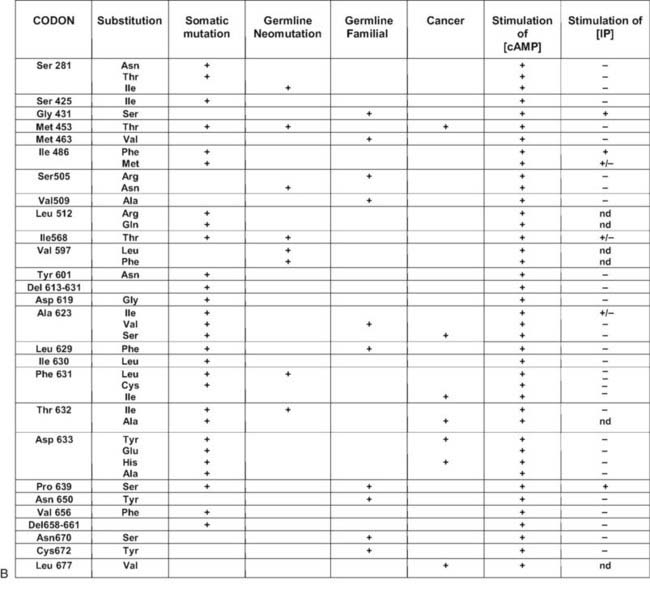
FIGURE 91-1. Schematic representation of the thyroid-stimulating hormone (TSH) receptor. A, The locations of known activating mutations are indicated.B, The nature of the mutations is indicated along with their origins (somatic, germline sporadic, germline familial, cancer) and effects on intracellular regulatory cascades.
(For a complete list of activating mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.)
The prevalence of hereditary toxic thyroid hyperplasia is difficult to estimate at the present time. It is likely that many cases have been (and still are) mistaken for Graves’ disease. This may be explained by the high frequency of thyroid autoantibodies (antithyroglobulin, antithyroperoxidase) in the general population. It is expected that wider knowledge of the existence of the disease will lead to better diagnosis. This is not a purely academic problem, in that presymptomatic diagnosis in children of affected families may prevent the developmental or psychological complications associated with infantile or juvenile hyperthyroidism. A country-wide screening for the condition has been performed in Denmark. It was found in 1 out of 121 patients with juvenile thyrotoxicosis (0.8%; 95% confidence interval [CI]: 0.02% to 4.6%), which corresponds to 1 in 17 patients with presumed nonautoimmune juvenile thyrotoxicosis (6%; 95% CI: 0.15% to 28.69%).21
SPORADIC TOXIC THYROID HYPERPLASIA
Cases of toxic thyroid hyperplasia have been described in children of unaffected parents.7,22–29 Conspicuously, congenital hyperthyroidism was present in most of the patients and required aggressive treatment. Mutations of one TSH receptor allele were identified in the children but were absent in the parents. Because paternity was confirmed by minisatellite or microsatellite testing, these cases qualify as true neomutations. When the amino acid substitutions implicated in hereditary and sporadic cases are compared, for the majority, they do not overlap (see Fig. 91-1). Although most of the sporadic cases harbor mutations that are also found in toxic adenomas, most of the hereditary cases have “private” mutations. Analysis of the functional characteristics of the individual mutant receptors in COS cells and the clinical course of individual patients suggests an explanation for this observation: “sporadic” mutations seem to have a stronger activating effect than “hereditary” mutations do. From their severe phenotypes, it is likely that newborns with neomutations would not have survived if not treated efficiently. On the contrary, from inspection of the available pedigrees, it seems that the milder phenotype of patients with hereditary mutations has only a limited effect on reproductive fitness. The fact that hereditary mutations are rarely observed in toxic adenomas is compatible with the suggestion that they would cause extremely slow tissue growth and, accordingly, would rarely cause thyrotoxicosis, if somatic. There is, however, no a priori reason for neomutations to cause stronger gain of function than hereditary mutations. Accordingly, an activating mutation of the TSH receptor gene has been found in a 6-month-old child with subclinical hyperthyroidism who presents with weight loss as the initial symptom.30
SOMATIC MUTATIONS: AUTONOMOUS TOXIC ADENOMAS
Soon after mutations of Gsα had been found in adenomas of the pituitary somatotrophs,31 similar mutations (also called gsp mutations) were found in some toxic thyroid adenomas and follicular carcinomas.32–35 The mutated residues (Arg201, Glu227) are homologous to those found mutated in the ras proto-oncogenes, that is, the mutations decrease the endogenous guanine triphosphatase activity of the G protein, thereby resulting in a constitutively active molecule.
Toxic adenoma was found to be a fruitful source of somatic mutations activating the TSH receptor, probably because the phenotype is very conspicuous and easy to diagnose. Whereas mutations are distributed all along the serpentine portion of the receptor36–42 and even in the extracellular aminoterminal domain,43 there is clearly a hotspot at the cytoplasmic side of the sixth transmembrane segment (see Fig. 91-1). (For a complete list of TSH receptor gene mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.) The clustering reflects the pivotal role of this portion of the molecule in activation mechanisms44–49 (see also Chapter 74).
Despite some dispute about the prevalence of TSH receptor mutations in toxic adenomas, which may be due to different origins among patients50 or different sensitivities of methods, the current consensus is that activating mutations of the TSH receptor are the major cause of solitary toxic adenomas and account for about 60% to 80% of cases.36,42,51–53 Contrary to initial suggestions, the same percentage of TSH receptor mutation is observed in Japan, an iodine-sufficient country with low prevalence of toxic adenomas.54 In some patients with multinodular goiter and two zones of autonomy at scintigraphy, a different mutation of the TSH receptor was identified in each nodule.55–58 This finding indicates that the pathophysiologic mechanism responsible for solitary toxic adenomas can be at work on a background of multinodular goiter. In agreement with this notion, activating mutations of the TSH receptor have been identified in hyperfunctioning areas of multinodular goiter.57,59–61 The independent occurrence of two activating mutations in a patient may seem highly improbable at first. However, the multiplicity of the possible amino acid targets for activating mutations within the TSH receptor makes this event less unlikely. It is also possible that a mutagenic environment is created in glands exposed to chronic stimulation by TSH in which H2O2 is produced.62,63 Finally, involvement of TSH receptor mutations in thyroid cancer has been implicated in a limited proportion of follicular thyroid carcinomas.61,64–69
STRUCTURE-FUNCTION RELATIONSHIPS OF THE TSH RECEPTOR, AS DEDUCED FROM ACTIVATING MUTATIONS
Most of the activating mutations of the TSH receptor have been studied by transient expression in COS or HEK293T cells. There is no guarantee that the mutants will function in an identical way in these artificial systems as they do in thyrocytes. Arguments have been obtained for such cell-type specific effects.70 In thyrocytes, a better relation has been observed between adenylyl cyclase stimulation and differentiation than with growth.70 However, the built-in amplification associated with transfection of constructs in COS or HEK 293T cells makes it possible to detect even slight increases in constitutive activity of TSH receptor mutants. An important observation has been that the wild-type receptor itself displays significant constitutive activity.38,71 This characteristic is not unique to the TSH receptor, but it is interesting to note that it is not shared by its close relatives, the luteinizing hormone/chorionic gonadotropin (LH/CG) receptor and the follitropin receptor (FSH). The effect of activating mutations accordingly must be interpreted in terms of “increase in constitutive activity.” Most receptor mutants found in toxic adenomas and/or toxic thyroid hyperplasia share common characteristics: (1) they increase the constitutive activity of the receptor toward stimulation of adenylyl cyclase; (2) with a few notable exceptions (see Fig. 91-1),72 they do not display constitutive activity toward the inositol phosphate/diacylglycerol pathway; (3) their expression at the cell surface is decreased (from slightly to severely); (4) most but not all of them keep responding to TSH for stimulation of cAMP and inositol phosphate generation, with a tendency to do so at decreased median effective concentrations; and (5) they bind 125I-labeled bovine TSH with an apparent affinity higher than that of the wild-type receptor.
No simple relationship exists between the position of the mutations or the nature of the amino acid substitution and their functional characteristics. Mutations found in transmembrane segments 1, 2, 3, 6, and 7 and the third cytoplasmic loop all have similar phenotypes; they involve amino acids belonging to all classes (charged, polar, hydrophobic), with substitutions not necessarily involving a shift to another class. Mutations involving Ile486 and Ile568 in the first and second extracellular loops, respectively, and Pro639 in transmembrane segment 6 are exceptional in that, in addition to stimulating adenylyl cyclase, they cause constitutive activation of the inositol phosphate pathway.
No direct relationship has been found between the level of cAMP achieved by different mutants in transfected COS cells and their level of expression at the cell membrane,73 which means that individual mutants have widely different “specific constitutive activity” (measured as the stimulation of cAMP accumulation/receptor number at the cell surface). Although this specific activity may tell us something about the mechanisms of receptor activation, it is not a measure of the actual phenotypic effect of the mutation in vivo. Indeed, one of the relatively mild mutations, observed up until now only in a family with HTTH (Cys672Tyr), is among the strongest according to this criterion. It would be logical to expect the best correlation to be found between the phenotype and the actual level of cAMP achieved, irrespective of the level of receptor expression. However, differences between the effects of the mutants in transfected COS or HEK293 cells and thyrocytes in vivo may render these correlations a difficult exercise.70
According to a current model of G protein–coupled receptor (GPCR) activation, the receptor would exist under at least two interconverting conformations: R (silent conformations) and R* (the active forms).44 The unliganded receptor would shuttle between both forms, the equilibrium being in favor of R. Binding of the ligand to the slit between the transmembrane segments (for biogenic amines) and/or the residues of the N-terminal segment or extracellular loops (for neuropeptides) is believed to stabilize the R* conformations. The resulting R-to-R* transition is supposed to involve a conformational change that modifies the relative position of transmembrane helices, which in turn would translate into conformational changes in the cytoplasmic domains interacting with trimeric G proteins. Seminal studies with the adrenergic receptor α1b have shown that a variety of amino acid substitutions in the C-terminal portion of the third intracellular loop lead to their constitutive activation.74 The observation that all amino acid substitutions at Ala293 were effective in activating the receptor led to the concept that the silent form of GPCRs would be submitted to a structural constraint requiring the wild-type primary structure of the third intracellular loop. This constraint could be released by a wide spectrum of amino acid substitutions in this segment.44,75
The observation that amino acid substitutions in a large number of residues scattered over the serpentine portion of the TSH receptor cause an increase in its constitutive activity is fully compatible with the above model and provided arguments for its extension (see Chapter 74). The fact that mutations in residues distributed over most of the serpentine portion of the receptor are equally effective in activating it (which does not seem to be a general characteristic in all GPCRs) suggests that the unliganded TSH receptor might be less constrained than others. The readily measurable constitutive activity of the wild-type receptor is compatible with this contention. Being already “noisy,” the TSH receptor would be more prone to further destabilization by a variety of mutations.
The precise effects of individual mutations in structural terms are beginning to emerge from analogies with the limited list of GPCRs whose tridimensional structure has been solved76 (see Chapter 74 for a discussion and for a model of activation of the TSH receptor).
FAMILIAL GESTATIONAL HYPERTHYROIDISM
Some degree of stimulation of the thyroid gland by human chorionic gonadotropin (hCG) is commonly observed during early pregnancy. It is usually responsible for a decrease in serum thyrotropin with an increase in the concentration of free thyroxine (T4), which remains within the normal range (for references, see 77). When concentrations of hCG are abnormally high, as in molar pregnancy, true hyperthyroidism may ensue. The pathophysiologic mechanism is believed to be promiscuous stimulation of the TSH receptor by excess hCG, as suggested by the rough direct or inverse relation between serum hCG and free T4 or TSH concentrations, respectively.77 A convincing rationale is provided by the close structural relationships and evolutionary origins of the glycoprotein hormones and their receptors.78
A new syndrome was described in 1998 in a family with dominant transmission of hyperthyroidism limited to pregnancy.79 The proposita and her mother had severe thyrotoxicosis together with hyperemesis gravidarum during the course of each of their pregnancies. When nonpregnant, they were clinically and biologically euthyroid. Both patients were heterozygous for a Lys183Arg mutation in the extracellular aminoterminal domain of the TSH receptor. When tested by transient transfection in COS cells, the mutant receptor displayed normal characteristics toward TSH. However, a convincing explanation for the phenotype was provided in that it showed higher sensitivity to stimulation by hCG than the wild-type TSH receptor.
The amino acid substitution responsible in these patients for promiscuous stimulation of the TSH receptor by hCG is surprisingly conservative. Also surprising is the observation that residue 183 is a lysine in both the TSH and LH/CG receptors. When placed on the three-dimensional structure of the hormone-binding domain of the TSH receptor,80 residue 183 belongs to one of the β sheets that constitute the surface of interaction with the hormones. Detailed analysis of the effect of the K183R mutation by site-directed mutagenesis indicated that any amino acid substitution at this position confers a slight increase in stability to the illegitimate hCG/TSH receptor complex.81 This increase in stability would be enough to cause signal transduction by the hCG concentrations achieved in pregnancy, but not by the LH concentrations observed after menopause. Indeed, the mother of the proposita remained euthyroid after menopause. This finding is compatible with a relatively modest gain in function of the Lys183Arg mutant upon stimulation by hCG.
In contrast to other mammals, humans and primates rely on CG for maintenance of the corpus luteum in early pregnancy. The frequent partial suppression of TSH observed at peak hCG levels during normal pregnancy indicates that evolution has selected physiologic mechanisms operating very close to the border of thyrotoxicosis. This finding may provide a rationale for the observation that in comparison with other species, the glycoprotein hormones of primates display lower biological activity because of positive evolutionary selection of specific amino acid substitutions in their α subunits.78 Up to now, no spontaneous mutation has been identified that would increase the bioactivity of hCG. An interesting parallel may be drawn between familial gestational hyperthyroidism and cases of spontaneous ovarian hyperstimulation syndrome (sOHSS).82,83 In sOHSS, mutations of the FSH receptor gene render the receptor abnormally sensitive to hCG. The result is recurrent hyperstimulation of the ovary, on the occasion of each pregnancy.
Loss-of-Function Mutations
Loss-of-function mutations in the TSH receptor gene are expected to cause a syndrome of “resistance to TSH.” The expected phenotype is likely to resemble that of patients with mutations in TSH itself. These mutations have been described early because of the prior availability of information on TSH α and β genes.84 Mouse models of resistance to TSH are available as natural (hyt/hyt mouse)85 or experimental TSH receptor mutant lines.86,87 It is interesting to note and contrary to the situation in humans (see below) that the thyroid of newborn TSH receptor knockout mice is of normal size. As expected, the homozygote animals displayed profound hypothyroidism. Their thyroids did not express the sodium-iodide symporter but showed significant (noniodinated) thyroglobulin production. From this information, one would expect patients with two TSH receptor–mutated alleles to exhibit a degree of hypothyroidism in accordance with the extent of loss of function, going from mild, compensated hypothyroidism to profound neonatal hypothyroidism with absent iodide trapping. Heterozygous carriers are expected to be normal or to display minimal increase in plasma TSH (see below).
CLINICAL CASES WITH THE MUTATIONS IDENTIFIED
A few patients with convincing resistance to TSH had been described before molecular genetics permitted identification of the mutations.88,89 The first cases described in molecular terms involved euthyroid siblings with elevated TSH.90 Sequencing of the TSH receptor gene identified a different mutation in each allele of the affected individuals, which made them compound heterozygotes. The substitutions occurred in the extracellular aminoterminal portion of the receptor (maternal allele, Pro162Ala; paternal allele, Ile167Asn). The functional characteristics of the mutant receptors showed that the paternal allele was virtually completely nonfunctional, whereas the maternal allele displayed an increase in the median effective TSH concentration for stimulation of cAMP production. Current knowledge of the tridimensional structure of part of the ectodomain of the receptor80,91 allows one to establish structure-function relationships for mutations affecting this portion of the receptor.92
A large number of familial cases with loss-of-function mutations of the TSH receptor have been identified in the course of screening programs for congenital hypothyroidism93–106 (Fig. 91-2). (For a complete list of TSH receptor gene mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.) Some of the patients displayed the usual criteria for congenital hypothyroidism, including high TSH, low free T4, and undetectable trapping of 99Tc. In some cases, plasma thyroglobulin levels were normal or high. Patients can be compound heterozygotes for complete loss-of-function mutations,94 or they may be homozygotes, born to consanguineous93 or apparently unrelated parents.100 In agreement with the phenotype of knockout mice with homozygous invalidation of the TSH receptor, patients with complete loss of function of the receptor display an in-place thyroid with completely absent iodide or 99Tc trapping. However, in contrast to the situation in mice, the gland is hypoplastic. Activation of the cAMP pathway, although dispensable for the anatomic development of the gland and thyroglobulin production, thus is absolutely required for expression of the sodium-iodide-symporter (NIS) gene and, at least in humans, for normal growth of the tissue during fetal life. This explains how in the absence of thyroglobulin measurements or expert echography, loss-of-function mutations of the TSH receptor may easily be misdiagnosed as thyroid agenesis.
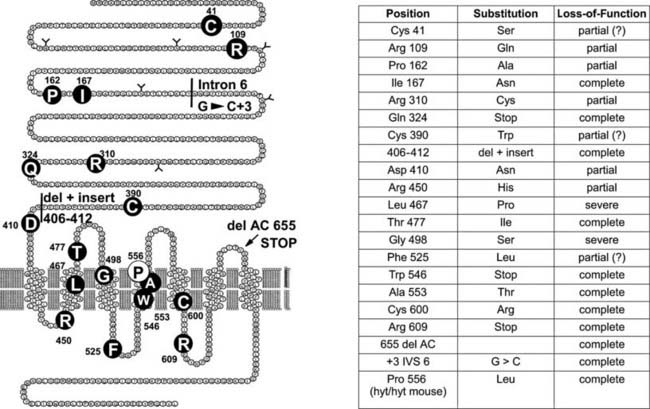
FIGURE 91-2. Loss-of-function mutations of the thyroid-stimulating hormone (TSH) receptor. The locations of known loss-of-function mutations are indicated (left) together with their nature and functional characteristics.
(For a complete list of loss of function mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


