FIGURE 127-1. Activation of steroid receptors. In the absence of ligand, steroid receptors (SR) reside in an inactive complex containing heat shock proteins (HSP) and other proteins. Upon steroid binding, the receptor undergoes a conformational change and dissociates from the HSP complex. The receptor/ligand complex dimerizes and interacts with hormone responsive elements (HRE) in the promoter region of steroid-responsive genes. The steroid receptor dimer at the HRE is thought to recruit coactivator proteins that in turn recruit other factors that together induce gene transcription.
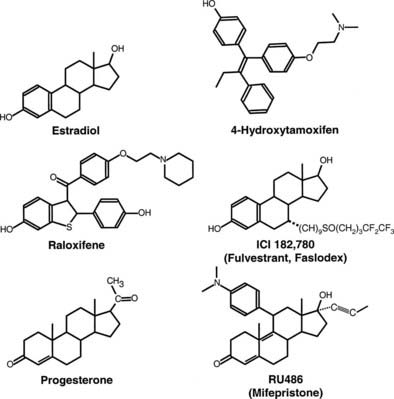
FIGURE 127-2. Structure of estrogen receptor (ER) and progesterone receptor (PR) agonists and antagonists. 17β-estradiol is the natural ER agonist. 4-Hydroxytamoxifen, raloxifene, and ICI 182,780 (fulvestrant, Faslodex) are estrogen receptor antagonists. Progesterone is the natural PR agonist, and RU486 (mifepristone) is an antagonist of PR action.
Structure of Estrogen and Progesterone Receptors
ESTROGEN RECEPTOR-α
The structure of the human estrogen receptor-α (ERα) is shown in Fig. 127-3. The amino terminal region (A/B) contains one of the transcriptional activation functions of ERα. This activity, originally termed TAF1,11 has been renamed activation function 1 (AF1). The A/B region in ERα is substantially shorter than that of other steroid receptors such as the PR shown in Fig. 127-4. Adjacent to the A/B region is the DNA binding domain (C), the most highly conserved domain in steroid receptors. This region contains two zinc finger motifs, each of which contains four cysteines that coordinately bind Zn++. The consensus DNA response element for the estrogen receptor is an inverted palindrome separated by three nucleotides (AGGTCAnnnTGACCT), although natural response elements may differ substantially from this consensus. The D or hinge region is poorly conserved among steroid receptors but contains a nuclear localization signal.12 The E region is required for hormone binding and contains dimerization domains,13 a second activation function (AF2), and sites for interaction with other proteins, including hsp9014 and a variety of co-activators. The estrogen receptor contains an additional region, F, at the carboxyl terminus.
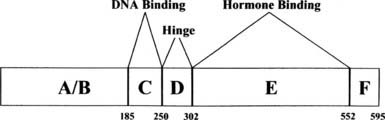
FIGURE 127-3. Structure of estrogen receptor-α. A through F represent the previously defined regions of the receptor described in the text.5 The numbers denote the amino acids at the boundaries of the regions.
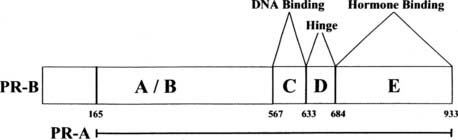
FIGURE 127-4. Structure of progesterone receptor A and B forms. The progesterone receptor (PR), like the estrogen receptor (ER), contains modular domains A through E with corresponding functions. The numbers denote the amino acids at the boundaries of the regions, with 1 corresponding to the first amino acid of the PR-B sequence.
ESTROGEN RECEPTOR-β
For many years, it was thought that there was only one estrogen receptor. However, a second estrogen receptor was cloned from a prostate cDNA library in 1996.15 The classical estrogen receptor described above therefore was renamed ERα, and the more recently discovered estrogen receptor is referred to as ERβ. This second estrogen receptor is encoded by a separate gene. It shares roughly 84% homology with ERα in the DNA binding domain, but only 58% in the E region and less in the other domains. In humans, five ERβ isoforms result from alternative splicing of the last coding exons.16 Studies have demonstrated functional differences for some of these isoforms, with ERβ2 lacking the ability to bind estradiol, and ERβ4 and ERβ5 heterodimerizing with ERβ1 and enhancing the latter’s transcriptional activity. In addition to forming homodimers and ERβ isoform heterodimers, ERβ can heterodimerize with ERα, with resultant activity intermediate between that of ERα and ERβ homodimers.17 Cellular responses to estrogens therefore are dependent on the relative abundance of ERα, ERβ, and its splicing isoforms.
PROGESTERONE RECEPTORS
Shown in Fig. 127-4 are the structures of the PR-A and PR-B forms of the human PR. Although there is only one gene for the PR, most species (with the exception of rabbit) express two forms of the receptor.18–20 Multiple mRNAs are produced from the progesterone receptor gene, and some of these lack the 5′ region that encodes the portion unique to the B receptor.21 The shorter form is produced by initiation of translation at an internal AUG.22 Thus, the two forms have identical hormone binding and DNA binding domains, but the shorter A form lacks the first 164 amino acids of the B form. The structure of the DNA binding domain is similar to that of the ER. The consensus DNA binding site (AGAACAnnnTGTTCT) for PR is an inverted palindrome separated by three nucleotides.23 As is true for estrogen response elements, progesterone response elements can differ substantially from the consensus sequence, and promoters of target genes may have multiple response elements and/or half-sites that contribute to the activity. Androgen receptors, glucocorticoid receptors, and mineralocorticoid receptors also bind to the PR consensus sequence.23 Although some hormone response elements are found near the promoter, some are >10 kb upstream and others are found within the transcribed region.24 PR binds to some target genes through protein-protein interactions, or through a combination of DNA and protein interactions.
The hinge region (D) of PR contains a nuclear localization sequence that is responsible for constitutive nuclear localization of the receptor.12 The E region contains the hormone-binding region, an AF2 function, and sites for dimerization and interaction with other proteins. Unlike ER, PR lacks an F region; deletion of even a few of the carboxyl terminal amino acids of PR results in loss of hormone binding.
The aminoterminal domains of PR-A and PR-B also contain an activation function, AF1. The relative importance of AF1 and AF2 in transcriptional activation depends upon the cell type and the promoter sequence. In many cases, the two act synergistically. An additional activation region, termed AF3, is found in the region unique to PR-B.25
Co-Activators and Co-Repressors
CO-ACTIVATORS
Until recently, the molecular basis for hormones bound to receptors acting as agonists or antagonists was unknown. Similarly, the means by which the activation functions enhanced transcription was, for the most part, unknown. The discovery of co-activators and co-repressors that regulate the activities of transcription factors has provided insight into these issues. Co-activators typically are recruited to active forms of transcription factors and enhance target gene transcription, whereas co-repressors bind to inactive forms of transcription factors, thereby repressing transcription. Large numbers of co-activators and co-repressors (>300) have been identified,26 and their abilities to regulate transcription are target gene specific.
In the case of steroid receptors, the activating signal is typically binding of agonist. Steroid receptors themselves have no enzymatic activity. Rather, they act as a platform or scaffold at specific sites within chromatin to recruit a series of proteins and protein complexes that have enzymatic activities. These proteins modify chromatin proteins and other proteins associated with chromatin to facilitate or prevent transcription of target genes. Co-activators for steroid receptors were originally sought as proteins that bind to the hormone binding domain only in the presence of agonist. The first nuclear receptor co-activator to be identified, steroid receptor co-activator-1 (SRC-1), is one of three related co-activators termed p160 co-activators.27 The p160 co-activators and many of the other receptor co-activators contain LXXLL (L = leucine, X = any amino acid) sequences, a motif that has been implicated in the binding of co-activators to steroid receptors,28 as well as in other protein-protein interactions. Many of the co-activators are large and contain multiple protein-protein interaction sites, as well as regions that encode enzymatic activities. Others appear to serve as scaffolds that recruit additional proteins to gene regulatory regions. SRC-1 has numerous protein-protein interaction sites and also is a histone acetyltransferase (HAT).29 Acetylation of histones by HATs alters their interaction with DNA and as a result relaxes chromatin structure for more efficient transcription. Although histones are substrates for HATs, some HATS also acetylate transcription factors, including p53 and some of the steroid receptors.30
Co-activators form complexes with other proteins, and recent data on isolation of co-activator complexes and on the kinetics of association of co-activator proteins with receptors on promoters suggest that groups of proteins are recruited as complexes through one or more proteins in the complex that interact directly with the receptor.31 In the case of SRC-1, the region that contains intrinsic HAT activity also binds another histone acetyltransferase, pCAF (p300/CBP-associated factor). Additional p160 interacting proteins include the CREB binding protein (CBP) co-activator, and the histone methyltransferases, CARM1 and PRMT1.32 In addition to the complexes that contain p160 proteins, several complexes are recruited to the promoters that modify chromatin and/or interact with and modify basal transcription factors. These include SWI/SNF complexes that mediate ATP-dependent chromatin remodeling, the TRAP/mediator complex that enhances recruitment and phosphorylation of RNA polymerase II,33 kinases such as cyclin T/Cdk9 (P-TEFb), and cyclin A2/Cdk2, and components of the proteasome pathway that facilitate gene expression.
More than 100 co-regulators have been implicated in human diseases via their mutation, overexpression, or underexpression, or because of polymorphisms in co-regulators linked to human pathology. For instance, overexpression of co-activators has been linked to carcinogenesis, tumor progression, and altered responses to endocrine-based therapeutics such as tamoxifen resistance in breast cancer.
CO-REPRESSORS
The steroid receptor family co-repressors were initially identified as proteins that interact with two steroid receptor family members (thyroid hormone receptor and retinoic acid receptor) that repress basal transcription of their target genes. The co-repressors bind to these proteins in the absence of ligand but are released when the receptors bind to their cognate agonist ligands. Two of the most extensively characterized co-repressors, NCoR (nuclear receptor co-repressor34) and SMRT (silencing mediator of retinoic acid and thyroid hormone receptors35), bind to unliganded retinoic acid receptor and thyroid hormone receptors and recruit multiprotein complexes that contain histone deacetylases.36 Deacetylation of the histones results in a more compact and therefore less accessible chromatin structure. With respect to steroid receptors, NCoR and SMRT originally were thought to be important only for the actions of pharmacologic steroid receptor antagonists. Several studies demonstrate that SMRT and NCoR bind preferentially to antagonist-rather than agonist-bound ER and PR in vivo.37–39 Overexpression of these proteins reduces the partial agonist activity of 4-hydroxytamoxifen bound to ER and the partial agonist activity of RU486 bound to PR. Hence, the prevailing model is that this class of proteins is clinically important in situations where receptor antagonists are administered. However, recent evidence suggests that the SMRT corepressor is important for E2-dependent expression of some ER target genes.40 This effect is tissue and gene specific and is consistent with other examples of co-activators repressing and co-repressors stimulating gene expression.41
Estrogen Receptor Action
The group of hormones referred to as estrogens are steroids that play important roles during development and various aspects of reproduction, but that also exert significant biological effects in the brain and in the cardiovascular and skeletal systems. The ability of estrogens to increase uterine size through epithelial cell proliferation and accumulation of water has long been used to monitor estrogen action in animal studies.42 Likewise, the effects of estrogen in the mammary gland, where they induce ductal growth and branching and formation of terminal end buds,43,44 and in the skeleton, where they promote skeletal maturation and maintenance of bone mineral density, are well recognized.45 Differences in the incidence of cardiovascular disease in premenopausal women compared with men suggest that estrogens may be cardioprotective, and evidence suggests that estrogens reduce the levels of serum cholesterol and have positive effects on serum lipoprotein profiles.46 This view, however, is at least partially contradicted by the results of the Women’s Health Initiative Study, which reveals a small but significant increase in cardiovascular events in postmenopausal women receiving hormone replacement therapy.47,48 Some but not all evidence indicates that estrogen action in the central nervous system is associated with protection against cognitive aging and a reduction in the risk for Alzheimer’s disease.49,50
With the finding that there are at least two estrogen receptors and that these receptors can be activated in the absence of hormone, simple elimination of estrogens is insufficient to assess the biological role of a specific receptor in vivo. Mice lacking ERα (known as ERKO) generated via disruption of the ERα gene by homologous recombination in embryonic stem cells have been used to evaluate the role of this receptor in vivo.44,51 Both male and female mice lacking ERα are viable; the females are infertile, whereas the males exhibit reduced fertility. Although prenatal reproductive tract development proceeds normally, several reproductive tract abnormalities, including hypoplastic uteri and hyperemic ovaries lacking corpora lutea, have been identified in the adult animals. Moreover, these animals fail to display characteristic responses to estradiol, including increases in uterine wet weight and vaginal cornification. The animals also exhibit reduced bone mineral density and elevated levels of gonadal steroids, indicating feedback control at the level of the hypothalamus, and the pituitary is absent.45 Thus, it appears that these actions of estrogen are mediated by ERα, and that ERβ cannot act as a substitute.
Mice lacking ERβ due to homologous recombination technology are viable and develop normally, and as might be expected from differences in the expression patterns of ERα and ERβ, the phenotype of ERβ knock-out (βERKO) mice is distinct from that of ERKO mice.44,52 Female βERKO mice have reduced fertility as the result of compromised follicular maturation, as evidenced by sparse corpora lutea; this appears to be due to a defect intrinsic to the ovary, instead of hyperstimulation of the ovary by luteinizing hormone produced by the pituitary, as is the case for ERKO animals. Male βERKO mice are fertile but exhibit signs of prostatic hyperplasia.53 Only female βERKO mice exhibit a bone phenotype consisting of increased bone length and cortical bone mineral content.45 Taken together, analyses of ERα and ERβ knockout mice indicate that the two receptors play different biological roles, and that the receptors are unable to functionally substitute for one another. Thus the overall effects of estrogens result from a range of activities mediated by two distinct receptors, as well as from the nongenomic effects of these ligands (see below).
ER Agonists and Antagonists
Agonist binding to ERs induces conformational changes that favor dissociation from the heat shock proteins, receptor dimerization, and binding to DNA. Synthetic compounds that compete with estradiol for binding to the ER and antagonize its activity have been developed; these have been useful both for understanding ER function and clinically in the treatment of breast cancer.
Antiestrogens for ERα have been separated into two classes based on their activities (reviewed in Macgregor and Jordan54). Whereas the type I antiestrogens have mixed estrogen/antiestrogen activity, the type II compounds are pure antagonists. Tamoxifen and related derivatives containing the core triphenylethylene structure and the benzothiophene, raloxifene, are examples of type I antiestrogens (see Fig. 127-2). These compounds induce receptor dimerization and DNA binding. In some cases, they produce transcriptionally inactive complexes, whereas in others, they function as weak agonists; the nature of the response is cell, tissue, and target gene specific. For example, tamoxifen exerts estrogen-like activity in the uterus but antiestrogen effects in the normal breast and many breast tumors.55 Although type I antagonists are incapable of inducing AF2 function, they do not block AF1 activity. For this reason, under conditions in which AF1 is active, type I antagonists can function as partial agonists. Conversely, in contexts where AF2 function is predominant, they are effective antagonists. The differences in responses between the agonists and the type I antagonists correlate with the ability of the agonist-bound receptor to efficiently recruit p160 co-activators, and antagonist-bound receptors have a greater propensity to recruit co-repressors. A comparison between the x-ray crystallographic structure of the ER ligand binding domains containing estradiol or type I antagonists reveals that raloxifene binding56 or 4-hydroxytamoxifen binding57 displaces a critical α helix, helix 12, into a position that occludes the interaction of p160 co-activators with the receptor’s co-activator binding surface. That co-repressors play an active role in repressing transcription by antagonist-bound receptors is supported by the finding that under conditions where co-repressor levels are low, type I antagonists can function as agonists.38,39,58
Type II antagonists typified by ICI 164,384 and ICI 182,780 are 7α-substituted derivatives of estradiol that generally are considered to not possess agonist activity.54 These antagonists have been shown to promote DNA binding. However, cystallographic analyses of ERβ liganded by ICI 164,384 reveal a structure in which the co-activator binding groove of the receptor is not formed. Moreover, these antagonists promote the rapid degradation of estrogen receptors, and this likely is important for the pure antagonist effects of these compounds.59
Most of the work characterizing ER antagonists has centered on the responses of ERα. Studies of ERβ reveal that there are differences in the responses of ERα and ERβ to antiestrogens. Whereas agonists typically function to stimulate the transcriptional activity of both receptors, although with somewhat different efficacies, and the type II antagonists act as pure antagonists for both, the type 1 antagonists for ERα appear to act as pure antagonists of ERβ, as measured on estrogen response element (ERE)-containing reporter genes.60
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


