FIGURE 93-1. Estimated proportional carriage of thyroxine (T4) on the three major plasma binding proteins is shown for euthyroid subjects with normal or variant T4 binding proteins. In the face of normal free T4 and thyroid-stimulating hormone (TSH) concentrations, the concentration of total T4 can vary from about 25 nmol/L in total TBG deficiency to about 1800 nmol/L in the Pro218 variant of familial dysalbuminemic hyperthyroxinemia (FDH) described in several Japanese kindreds.
Concentrations of the binding proteins can vary independent of thyroid status. When the concentration of TBG changes, the total serum T4 and T3 concentrations tend to alter to restore the preexisting concentration of free hormone, as determined by the set point of the feedback relationship with TSH. Hence, in theory, free hormone estimates will give a more accurate reflection of thyroid status than will measurement of total hormone. However, sample storage and dilution and the presence of competitors can alter the relationship between free and total hormone concentration, so that an analytically correct free hormone estimate may not reflect the in vivo situation (see below).
Characteristics of Hormone Binding
The definition of a number of terms assists the understanding of thyroid hormone binding. Capacity expresses the molar concentration of a specific class of ligand-binding site; if one binding site is present per protein molecule, capacity and protein concentration will be identical in molar terms. When about half the binding sites are empty, the free and hormone concentrations will change to about the same extent (i.e., the free fraction will show little change). However, as the total concentration of ligand approaches the binding protein capacity, the free hormone concentration will rise disproportionately, as occurs in thyrotoxicosis as the total T4 serum concentration approaches the capacity of TBG.6
Proportional carriage, the distribution of total hormone between a number of heterogeneous binding proteins, is influenced by the concentration of binding protein, the affinity of binding, and the free hormone concentration, but it is not directly influenced by the total hormone concentration.7 In a heterogeneous mixture of binding proteins, as in serum, proportional carriage can change as the free hormone concentration alters in response to dilution or hormone loading (see below).
Free fraction describes the percentage of the total hormone that is unbound. When serum that contains a highly bound ligand is progressively diluted, bound hormone will dissociate, so that the free hormone concentration at first is well maintained, with an increase in free fraction.
Occupancy, the proportion of a particular class of binding site that is filled with hormone, is a direct function of the free hormone concentration and is fundamental to the definition of affinity or kD. TBG is normally about one third occupied by T4; TTR is <1% occupied by T4, and albumin shows negligible occupancy (Table 93-1).
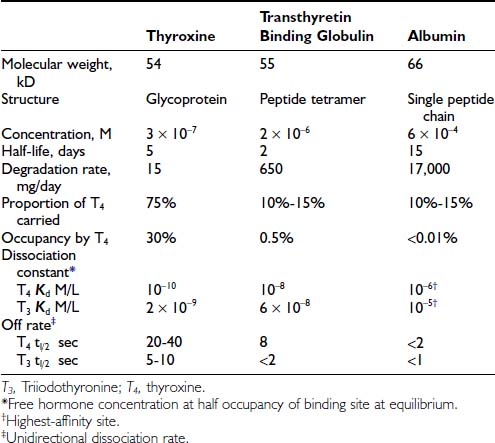
Affinity or dissociation constant (kD, mol/L) describes the free hormone concentration at which a particular binding site is half occupied. The association constant (kA, L/mol) is the inverse of kD. (kA is a theoretical concept of the number of liters at which 1 mole of ligand would occupy half the sites on 1 mole of binding protein.) By definition, a binding protein is 50% occupied when the free hormone concentration is the inverse of kA.
Dissociation rate (TΩ, sec) or rate constant (sec-1) defines the rate of unidirectional dissociation or delivery of hormone from a binding site.4 The unidirectional maximum rate of hormone delivery is relevant under non–steady state conditions, as, for example, when free hormone is rapidly removed from the circulation during tissue transit. The dissociation rate, as well as kA and kD, are highly temperature dependent. The free T4 fraction is higher at 37°C than at room temperature by a factor of up to two8 and dissociation of ligand is much faster.4
The reversible interaction between free T4 (fT4) and the unoccupied TBG-binding sites (uTBG) can be represented by the following:
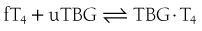
When represented in terms of the association constant, kTBG, this relationship becomes
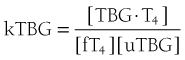
It follows that
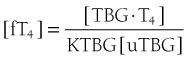
At half occupancy of the binding site, [TBG · T4] will equal [uTBG] and
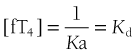
For the total hormone concentration to have a regulatory influence on tissue function, it is appropriate for the kD of the dominant binding protein and the physiologic concentration of free hormone to be of the same order, as is the case for the relationship between normal serum free T4 (10 to 25 pM) and the kD of TBG (100 pM). A wide disparity between the kD of the dominant binding protein and the free hormone concentration would require relatively large changes in total hormone concentration to achieve regulatory variation. In effect, TBG stabilizes the tissue distribution of T4, while allowing normal regulatory variation over an approximately twofold range of total hormone concentration. In the absence of TBG, a greater proportional change in total hormone concentration would be necessary to achieve regulatory variation.
Free Hormone Hypothesis
According to this hypothesis, it is the free or unbound equilibrium concentration of a hormone that determines biological activity. The validity of this hypothesis, which generally is well sustained for the thyroid hormones, has been analyzed in detail.9,10 Earlier liver perfusion experiments that showed the loosely albumin-bound moiety of the total circulating hormone pool to be virtually as readily available as the free hormone11 have now been refuted.9 No conclusive evidence suggests that any particular class of binding protein facilitates tissue uptake of thyroid hormones. When isolated rat liver was perfused with T4 bound to various normal and variant binding proteins,12 tissue uptake of T4 was proportional to the spontaneous dissociation of T4 from each protein.12
Under some circumstances, especially when capillary transit is slow, or when mixing of layers across the diameter of a vessel is incomplete, tissue uptake of hormone may be limited by dissociation of bound hormone.9 Under these circumstances, the local concentration of free hormone at a particular site may be lower than the equilibrium concentration. The albumin-bound moiety with the fastest rate of unidirectional dissociation (see Table 93-1) will then make a large contribution in replenishing the free concentration.9
As formulated by Mendel,9,10 the unmodified free hormone hypothesis will be valid when tissue uptake of hormone is limited by influx or elimination. When flow or dissociation is the limiting condition, for example, when flow is slow and clearance is rapid in a tissue such as the liver, the free hormone hypothesis still holds, with hormone dissociation as an additional critical variable.
Structure and Binding Characteristics of Normal and Variant Transport Proteins
The characteristics of the three normal major iodothyronine-binding proteins, thyroxine-binding globulin, transthyretin and albumin, are summarized in Table 93-1. The numerous structural variants of the three major thyroid hormone–binding proteins that have been described (Table 93-2) were initially recognized from the investigation of euthyroid subjects who showed markedly abnormal levels of total serum T4 or T3, but recently, additional variants have been identified from population screening and genetic studies. The total concentrations of serum T4 and T3 range widely in association with variant binding proteins, but none of the multiple hereditary binding alterations has been shown to confer any advantage or disadvantage, or to disturb thyroid hormone action, unless thyroid disease is associated. Plasma TSH concentrations remain normal, as do free T4 and T3 concentrations, provided the method of estimation is free of artifact. In general, TBG abnormalities tend to affect T4 and T3 similarly; by contrast, some albumin variants lead to selective abnormalities of T4 or T3 binding.
Variants of iodothyronine-binding proteins have attracted the attention of clinicians and basic scientists for numerous reasons, including the following:
In contrast to the multitude of clearly defined plasma protein–binding variants, the possibility of genetically determined abnormalities of cell membrane transport, cytoplasmic binding, or deiodination of iodothyronines is less clearly defined (see below).
THYROXINE-BINDING GLOBULIN (TBG)
TBG is a single polypeptide chain α globulin, with molecular weight of about 54 kD synthesized as a 415 amino acid protein.13–15 The first 20 amino acid residues of the TBG peptide are hydrophobic in nature and probably represent the signal peptide, which is removed in the endoplasmic reticulum, leaving a mature protein of 395 amino acids in a single chain with a molecular weight of about 44 kD. Multiple glycosylation sites allow an average of 10 terminal sialic acid moieties. The carbohydrate portions of TBG influence protein half-life in blood, stability in vitro, and microheterogeneity on electrophoresis, with only minor effects noted on immunoreactivity or T4 binding. Although TBG is stable in stored serum at 4°C, it gradually loses its binding affinity for T4 at 37°C or above. Differences in the rate of loss of binding affinity at raised temperature have been important in identifying TBG variants. Of particular interest are a variant with markedly increased heat stability (TBG-Chicago)16 and a variant with an extremely heat-labile protein with an abnormally high concentration of denatured TBG and subnormal total T4 (TBG-Gary).17
The amino acid sequence of human TBG shows homology with rat TBG (70%), human cortisol-binding globulin (55%) and members of the serum protease inhibitor family (SERPINS), which includes α-antitrypsin (53% homology) and a 1-antichymotrypsin (58% homology).18 The significance of the structural similarity between human TBG, CBG, and the SERPINS remains unclear, because the hormone-binding proteins do not exhibit antiprotease activity.
Susceptibility to cleavage and change in configuration may modulate hormone delivery from the SERPIN family of binding proteins. The study of Zhou et al.19 confirms that the binding of thyroxine to TBG can alter in response to changes in configuration of the binding protein. Using nonglycosylated recombinant human TBG, investigators reported that thyroxine is carried in a surface pocket and not within the beta barrel of the TBG molecule. With this structural model, conformational changes that result from relocation of a mobile peptide loop within the TBG molecule can favor binding or release of thyroxine. The demonstration of labile interaction between TBG and thyroxine raises the important possibility of modulated or tissue-specific delivery of thyroxine that could vary in response to changes in local pH, temperature, or redox status. The details of how local tissue factors may enhance or limit local hormone release from SERPIN binding proteins remain to be studied (see later section, “Function of Iodothyronine Binding Proteins”).
The normal concentration of human plasma TBG measured by radioimmunoassay is between 10 and 30 mg/L (0.2 to 0.6 µmol/L). TBG is normally 20% to 40% occupied by T4 and <1% occupied by T3. Occupancy may increase markedly in hyperthyroidism owing to increased total T4 and decreased TBG concentrations, leading to a disproportionate rise in free T4 relative to total T4.6 The T4-binding affinity (kD), is about 50 pmol/L at 37°C, consistent with the estimate that TBG is approximately 30% occupied by T4 at the normal free T4 concentration of about 20 pmol/L.20
HEREDITARY TBG VARIANTS
The single 8 kilobase human TBG gene has been localized to the long arm of the X chromosome at site Xq21-q22.14 Male hemizygotes who express a single mutant allele can show one of three variant phenotypes for T4 binding to TBG: increase, decrease, or absolute deficiency. Despite the presence of two X chromosomes, normal females have TBG levels similar to those of males. Females heterozygous for complete TBG deficiency usually show less than the anticipated 50% reduction in serum TBG, a phenomenon attributed to selective inactivation of the mutant allele.14,15 However, selective inactivation of the normal allele occasionally may result in females showing complete deficiency of TBG.21
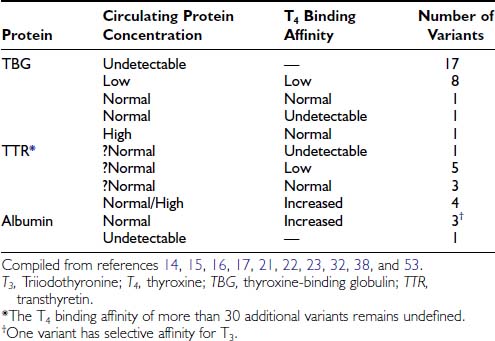
Multiple inherited TBG variants, often designated geographically, can result in partial or complete deficiency of immunoreactive TBG in serum. Of at least 24 known X-linked TBG mutants, 15 may cause complete TBG deficiency, and eight other variants are associated with subnormal concentrations of immunoreactive serum TBG, often with reduced affinity for T4.13–1522 The structural basis of numerous variants is summarized in Fig. 93-2. Several variants of TBG show decreased heat lability in vitro, which generally correlates with accelerated clearance in vivo. TBG deficiency (complete or partial) results from missense or nonsense mutations in the coding exons or in donor or acceptor splice sites.14,15 Thus, single nucleotide deletions or substitutions can lead to a frame shift with premature termination of translation, resulting in a truncated protein that is retained and degraded intracellularly.13–15 In some reports no mutations could be demonstrated in the TBG gene.23
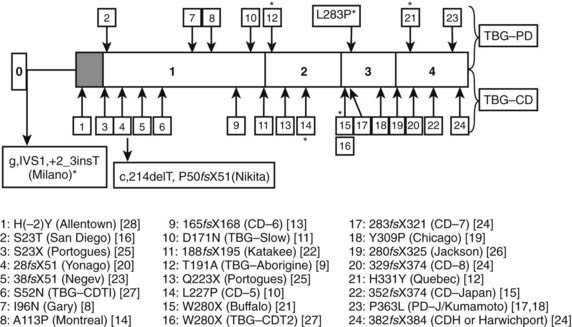
FIGURE 93-2. Summary of reported thyroxine-binding globulin (TBG) deficiency mutations. The numbered panel designates the five exons of TBG. Mutations above that panel refer to partial deficiency of TBG, and those below refer to total TBG deficiency. Polymorphisms are designated by an asterisk. Below the figure, details of each mutation are given, together with geographic designation and literature reference, as in Mannavola et al.14
(From Mannavola et al., 2006.14)
In general, the various methods for serum free T4 estimation, as well as binding corrections based on T3–uptake measurements, give a useful semi-quantitative correction for TBG abnormalities, whether hereditary or acquired.
In total TBG deficiency, total T4 is about 25 nmol/L (see Fig. 93-1), associated with normal free T4 and TSH. By contrast, in hemizygous TBG excess, the total T4 concentration is typically over 200 nmol/L, of which over 80% is carried on TBG (see Fig. 93-1).
From newborn screening studies, the prevalence of complete TBG deficiency in males is about 1 : 5000, with 1 : 15,000 showing complete deficiency,15 but marked ethnic differences are noted in the frequency of hereditary TBG deficiency, with complete deficiency being highest in the Japanese.14 Diminished TBG binding of T4 is especially prevalent in Australian aborigines, up to 30% of whom have subnormal serum concentrations of total T4, associated with subnormal serum concentrations of an abnormally heat-labile TBG that shows subnormal affinity for T4.24 Owing to a very high gene frequency in this population, the pattern of inheritance was initially thought to be autosomal dominant.25 Abnormal heat lability at 37°C was found in both male and female subjects from affected families, but the pattern of intermediate heat lability was found exclusively in female subjects,26 demonstrating that inheritance must be X-linked (Fig. 93-3). Hereditary TBG excess, probably due to gene duplication,27 appears to have a prevalence of about 1 : 25,000 in newborn males.15 The binding of T4 to TBG in inherited X-linked TBG excess is indistinguishable from the common type of TBG. In contrast to the albumin variants, no known TBG mutant shows increased T4-binding affinity.
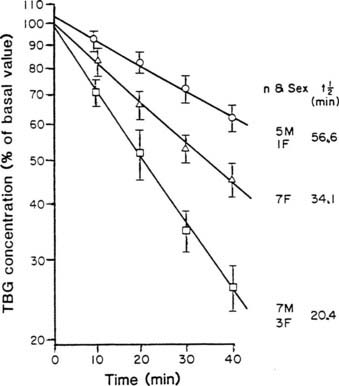
FIGURE 93-3. Heat stability of thyroxine-binding globulin (TBG) at 56°C in sera from Australian aborigines. Both male and female subjects showed either normal (upper line) or markedly reduced stability (lower line). No male subject showed the intermediate affinity (middle line) that demonstrates the heterozygous state, thereby confirming X-linked inheritance.
(From Refetoff S, Murata Y: X-chromosome-linked inheritance of the variant thyroxine-binding globulin in Australian Aborigines, J Clin Endocrinol Metab 1985;60:356-360.)
ALBUMIN
Human serum albumin, a highly conserved 66 kD nonglycoprotein,28 has a molar plasma concentration of approximately 600 µmol/L, corresponding to about 40 g/L. As well as being the principal carrier of numerous hydrophobic compounds in serum, albumin binds T4 in its region 2, with an affinity about four orders of magnitude less than that of normal TBG. Albumin normally carries 10% to 15% of circulating T4, but the proportion of albumin occupied by T4 is less than 0.002%.
HEREDITARY ALBUMIN VARIANTS
Hyperthyroxinemia can result from variant albumins with increased affinity for T4 or T3, the total albumin concentration being normal (Table 93-3). In familial dysalbuminemic hyperthyroxinemia (FDH), the total T4 concentration in affected individuals is about 200 nM,29,30 with over 50% of T4 carried on the variant albumin (see Fig. 93-1). FDH appears to be the most common hereditary T4-binding abnormality, with a prevalence as high as 1 : 1000 in some Latin American populations.31 As with other variants that show enhanced binding affinity or capacity, the increased concentration of total circulating T4 appears to be an appropriate response to maintain a normal free T4 concentration in feedback relationships with TSH.30
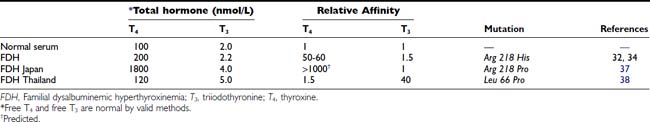
In FDH, Scatchard analysis of albumin binding shows two T4-binding sites: a normal site with kD 4.3 mmol/L and an abnormal site with 50- to 100-fold higher affinity, kD 50 nmol/L.32 The capacity of the higher-affinity T4-binding site is approximately 200 µmol/L, suggesting that relative to the molar concentration of albumin, at least one third of albumin molecules have the abnormal binding site.32 As a result, the occupancy of albumin by T4 increases about fivefold to about 0.01%. The common FDH variant is due to Arg-His substitution at position 218 of human albumin,33 with a T4 affinity 65-fold greater than normal,34 similar to the affinity reported for the natural protein more than a decade before.32 Kinetic studies in vivo show altered distribution of T4 in favor of the extracellular compartment and a reduced metabolic clearance rate of T4.35
In FDH, because of a markedly increased affinity of the variant protein for numerous T4-analogue tracers, serum free T4 estimated by early analogue-tracer methods yielded results suggestive of thyrotoxicosis.36 Greater albumin binding of tracer in FDH samples than in normal serum standards made less tracer available for binding to the assay antibody; the spurious decrease in bound counts resulted in a falsely high free T4 estimate.
A related autosomal dominant albumin variant at the same site (Arg 218 Pro), so far reported only in the Japanese,37 shows an even higher selective T4 affinity than the common FDH phenotype found in Caucasians. In euthyroid subjects with normal TSH, total T4 is almost 20-fold elevated at about 1800 nmol/L (see Fig. 93-1), whereas total T3 is only about twofold elevated. Free T4 estimates using analogue tracer also show spurious elevations in this variant.37
To explain the mechanism of increased T4 affinity in both types of FDH, it has been suggested that the guanidine group of arginine 218 normally may give an unfavorable binding interaction with T4. Histidine and proline, which lack the guanidine group, allow higher-affinity binding than is seen with wild-type albumin.37
A recent report described a mutation of the albumin gene at a different site (Leu 66 Pro) with selective affinity for T3 rather than T4.38 Eight euthyroid members of a Thai family showed total T3 levels of 4 to 8 nmol/L (normal, 1.0 to 2.6) when measured by radioimmunoassay with the use of I125 T3. Free T3 and free T4 were normal. This mutant albumin was estimated to have a 40-fold higher T3 affinity than normal albumin, but only a 1.5-fold higher affinity for T4. However, spuriously low total T3 values were found when T3 conjugates were used as tracer in enzyme-linked immunosorbent (ELISA) assays.38 The conjugate tracer, linked to alkaline phosphatase or to peroxidase, showed less binding to the variant albumin than to the natural protein, making more tracer available for binding to antibody in sample than in standard, the opposite artifact to that found with analogue-tracer free T4 estimates in the common type of FDH.
Only a few cases of total hereditary analbuminemia have been described in man, but in one kindred,39 evidence of mild TSH excess was found, consistent with impaired thyroid hormone delivery. In contrast, the Nagase strain of analbuminemic rat showed no evidence of any abnormality of thyroid hormone action or distribution.40
TRANSTHYRETIN
Transthyretin (TTR, previously known as prealbumin), a protein of approximately 55 kD that circulates in the serum of a wide range of vertebrates, is a tetramer consisting of four identical polypeptide chains held together by noncovalent bonds. Each monomer is a 127 amino acid chain regulated by a single gene on chromosome 18. The tetramer is symmetrical about a central cavity that completely penetrates the molecule and contains two T4-binding sites, one at each end of the central cavity.41
The normal serum concentration of TTR in healthy humans (2 to 8 µmol/L, 100 to 400 mg/L) can decrease rapidly during acute illness or malnutrition as a result of reduced hepatic synthesis.42 At normal concentrations, TTR is <1% occupied by T4, with a T4 affinity (kD ≈ 10 nmol/L) lower than TBG and higher than albumin. TTR has about 10-fold lower affinity for T3 than for T4. The liver is the principal site of synthesis of TTR, but the choroid plexus43 and the pancreatic islets44 are additional sites of TTR synthesis. In evolutionary terms, TTR synthesis at the choroid plexus long preceded the ontogeny of TTR synthesis in the liver.45 The T4-binding domain of TTR appears to have been conserved over the past 350 million years.45
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


