FIGURE 138-1. Schematic representation of the pathogenesis and clinical representations of testicular dysgenesis syndrome (TDS).
(Modified from Skakkebæk NE, Rajpert-De Meyts E, Jørgensen N, et al: Testicular cancer trends as “whistle blowers” of testicular developmental problems in populations, Int J Androl 30:198–205, 2007.)
Cryptorchidism
Cryptorchidism, or undescended testis, is one of the most common urogenital abnormalities in boys. It is mostly found isolated, but it can also occur in combination with other genital, urologic, skeletal, and neurologic malformations. Any disturbance in testis development and differentiation can compromise testis descent, and the severity of testicular dysgenesis is reflected in the severity of maldescent. Thus, an undescended testis is often indicative of some degree of testicular dysfunction, which may have long-lasting effects on especially semen quality and the risk of testicular cancer in adulthood.
CLASSIFICATION
Cryptorchidism is divided into a congenital and an acquired form, the distinction of which requires repetitive examination of boys during infancy and childhood. Synonymous terms for these two entities are primary and secondary cryptorchidism.8,9 Traditionally, cryptorchid testes are divided into nonpalpable and palpable testes, and synonyms used in literature are undescended testis, maldescensus testis, and retentio testis. During surgery, a nonpalpable testis may be revealed as being completely absent due to torsion or congenital atresia. Ideally, the precise position of the testis along its normal pathway of descent should be described in more detail: high/low abdominal testes, inguinal, suprascrotal, high scrotal, scrotal. Such a distinction would ensure a better basis for future studies about the long-term impact of treatment regimens and the disease per se on testis function. In rare cases, the testis can be found in an ectopic position outside its normal pathway of descent. Unilateral cryptorchidism occurs more frequently than bilateral cryptorchidism, and in only approximately 10% of orchidopexies, the testis is nonpalpable before surgery10 (Fig. 138-2). Retractile testes are considered a normal variant. However, in severely retractile cases or in children who are distressed during the physical examination, these testes may be difficult to distinguish from undescended testes. In addition, retractile testes have an increased risk to become truly cryptorchid over time, and their position needs to be followed.11
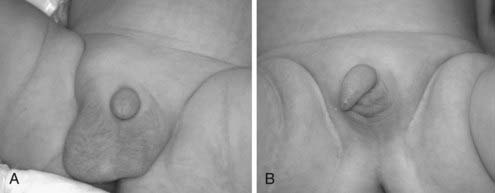
FIGURE 138-2. Infants with unilateral (A) and bilateral (B) cryptorchidism. It is important that the diagnosis is not based on the visual inspection but by a careful palpation of scrotum in warm and comfortable conditions.
Over time, many clinical classifications of testicular position have been used, which considerably impairs comparison of results across studies. Attempts have been made to facilitate standardization by measuring the distance between the top of the pubic crest and the middle of the testis after gentle traction. Distances of >4 cm in mature and >2.5 cm in premature boys are considered indicative of normal descent.12 However, this method has not found widespread application, since the measurements are highly dependent on age and size of the child.
Registry data, either of congenital malformations or retrospective analysis of hospital diagnoses after orchidopexy, may differ considerably in their selection criteria from prospective standardized studies, and the thoroughness of the report may be biased towards the more severe forms of testicular maldescent.13 Cumulative orchidopexy rates may also include cases of ascended testes and encompass changes in referral traditions and treatment regimens over time. Thus, reported prevalence rates may differ even within the same population.
The clinical examination can on occasion be technically difficult. It requires an experienced observer, warm and relaxed conditions, and may need to be repeated if the child is upset. Examination should not only be done in the supine position but also cross-legged or upright before a definite diagnosis is made. An experienced examiner may have a higher sensitivity for locating an undescended testis than ultrasound or other imaging techniques.14 The testis is brought into the lowest possible position along its normal pathway of descent by gentle, not forced, traction and should remain there for a while after exhaustion of the cremaster reflex. An asymmetric or hypoplastic scrotum may further suggest unilateral or bilateral cryptorchidism, respectively.
The position of the testis during infancy and childhood is not necessarily stationary, and there is a general consensus that boys should be examined repetitively as part of their standard health care program.8 A significant proportion of the testes in congenital cryptorchidism descend spontaneously during the first months of life.2 Testes, scrotally placed at birth, can also ascend during infancy and childhood,15 the etiology and consequences of which are yet unclear.
EMBRYOLOGY
The development of the testis and its descent from the abdomen into the scrotum is a highly complex process which is not yet completely understood. Numerous genetic and hormonal factors have been identified that play a crucial role.9 The development of the testis from the undifferentiated gonad commences around 5 to 6 weeks of gestation by migration of germ cells into the genital ridge and their differentiation into gonocytes.16 Genetic factors such as SRY, WT-1, SF-1, SOX9, Fgf9, and DAX1 contribute to the differentiation of cells from the coelomic epithelium into Sertoli cells. These aggregate around germ cells to form the primary testicular cords around 6 to 7 weeks. By the end of week 9, interstitial cells differentiate into Leydig cells. Leydig cells secrete testosterone (T), which induces male differentiation of the Wolffian duct into accessory reproductive organs in weeks 8 to 12 and masculinization of the external genitalia after conversion into dihydrotestosterone (DHT). They also secrete insulin-like hormone 3 (INSL3), which (at least in mice) induces male-like development of the gubernaculum. Secretion of antimüllerian hormone (AMH) by Sertoli cells from week 8 induces regression of the müllerian structures.
Testicular descent in humans occurs in two phases, the transabdominal phase and the inguinoscrotal phase, which are usually completed around gestational weeks 15 and 35, respectively.17 During the first phase, the testis is anchored at the internal inguinal ring by enlargement of the gubernaculum. In rodents, the masculinization of the gubernaculum is highly dependent on INSL3 and its receptor, leucine-rich repeat-containing G protein–coupled receptor (LGR8, RXFP2), whereas mutations of these genes in humans have only rarely been reported in large series of patients with cryptorchidism. The regression of the cranial suspensory ligament and the second phase of descent are highly dependent on androgens. The inguinoscrotal descent is therefore often compromised in hormonal disorders of the pituitary-gonadal axis or steroidogenesis and androgen-receptor mutations. The role of the genitofemoral nerve (GFN) and its neurotransmitter, calcitonin gene-related peptide (CGRP), in the inguinoscrotal phase of descent is less well established in humans than in rodents. However, the high prevalence of cryptorchidism in spina bifida and caudal regression syndromes indicates that GFN also plays a role in testis descent in humans.
In contrast to testicular descent, the differentiation of fetal gonocytes into spermatogonia and the increase in number of Leydig and Sertoli cells is a long process which is not completed at birth but continues during the postnatal period in humans for a few months. Differentiation of the first fetal gonocytes into spermatogonia begins at gestational week 13 to 15, with gradual down-regulation of stem-cell markers such as OCT-3/4, NANOG, TFAP2C, and KIT and appearance of germ-cell-specific proteins such as MAGEA4.18–25 During differentiation, germ cells migrate towards the basal lamina and appear as a dark spermatogonia in histologic sections.26 The increase in numbers of germ cells, Leydig cells, and Sertoli cells during the immediate postnatal period occurs in parallel with a brief surge of gonadotropins, testosterone, inhibin B, and AMH.27–29 This hormonal activation, also called mini-puberty, is thought to be essential for the final transformation of gonocytes or pre-spermatogonia into a dark spermatogonia.17
ETIOLOGY
Cryptorchidism can be caused by a multitude of genetic and hormonal disorders, as well as complex syndromes with midline and caudal defects. In addition, lifestyle factors and the environment may have adverse effects on testicular descent in humans.11 Disorders of the hypothalamic-pituitary-gonadal axis and testosterone biosynthesis have more frequently been reported in association with isolated cryptorchidism than genetic defects such as androgen-receptor mutations, polymorphisms of the CAG repeat of the androgen receptor, 5α-reductase deficiency, HOXA10 or INSL3 gene mutations, and polymorphism of the estrogen receptor alpha gene. However, there are familial cases of cryptorchidism, and a recent study estimated the relative risk of cryptorchidism to be increased to 6.9 if a brother and 4.6 if the father also had cryptorchidism.30
Epidemiologic studies have revealed a multitude of risk factors that are associated with congenital cryptorchidism.11 Among the most established risk factors are low birth weight, in particular being small for gestational age, and prematurity. Pregnancy complications such as placental insufficiency, preeclampsia, and maternal diabetes have also been associated with cryptorchidism in several studies. Even mild gestational diabetes appears to increase the risk of cryptorchidism in the offspring, potentially via reduced levels of maternal sex-hormone binding globulin (SHBG) and fetal hyperinsulinemia leading to an imbalance in the fetal estrogen-androgen action.31
Risk factors in maternal behavior, such as continuous smoking, drinking, and caffeine intake during pregnancy also seem to have adverse effects on the development and descent of the testis in their offspring,32–34 although these findings are still controversial.35 Some discrepancies in the results between studies are caused by the differences in the inclusion criteria related to mild and transient forms of cryptorchidism in some and only persistent or severe forms in others. These different types of cryptorchidism may not share the same etiology. Maternal smoking affected semen quality and testis volume of their adult offspring.36,37
Several epidemiologic studies suggest an increased prevalence of cryptorchidism and related urogenital malformations in specific professions such as farmers and males from geographic regions with high agricultural activity.38–40 In animal studies, prenatal exposure to many man-made chemicals with endocrine-disrupting properties can cause cryptorchidism,41,42 although the role of estrogenic compounds has recently been contested.43 Current evidence in humans is sparse, but there is emerging evidence that halogenated compounds such as polychlorinated compounds and polybrominated flame retardants may be associated with an increased risk of congenital cryptorchidism and occurrence of hypospadias.44–49 In addition, phthalate exposure of the fetus and infant was found to affect testicular hormonal function50 and reduce anogenital distance (AGD) as a sign of reduced virilization. Short AGD was at the same time linked to a high prevalence of cryptorchidism.51 In contrast to experimental animals, humans are exposed to a multitude of chemicals over the entire lifespan. First studies of so-called “cocktail effects” by exposing animals to low doses of chemicals suggest that this type of exposure may cause unexpected and potentially larger effects than anticipated.52,53 In the majority of cases with cryptorchidism, a distinct etiology cannot be ascertained, suggesting that genetic predisposition together with environmental and lifestyle factors may exert adverse effects.54
PREVALENCE
The reported prevalence of cryptorchidism varies considerably, depending on the type of study, geographic region, the selection, age, and ethnicity of participants, and the clinical classification. At birth, reported rates from prospective studies vary between 1.6% and 9%, if premature boys are also included, and between 1.8% and 8.4% for boys ≥2500 grams birth weight.2,11,13,55 Some countries, such as Denmark and Great Britain, have noted an increase of cryptorchidism over the past decades. A doubling of congenital cryptorchidism was also reported in the United States, Canada, and South America in the 1970s and 1980s. In contrast, orchidopexy data from registries suggest a decrease since the 1990s. In general, rates of cryptorchidism reported in registries tend to be lower than from prospective studies because ascertainment and registration traditions differ from systematic studies. For orchidopexies, registries may also reflect changes in treatment traditions and surgical referrals.
Owing to spontaneous postnatal descent of retained testes during the first months of life, prevalence rates drop to between 0.9% and 1.8% at 3 months of age. During childhood, retractile testes and ascensus testis become more common, and reported prevalence data from prospective studies increase up to 7%. The majority of ascended testes seem to descend spontaneously during puberty, thus the age and pubertal stage at examination has a major impact on reported rates.15
TREATMENT
There have been two treatment modalities available for cryptorchidism: hormonal or surgical treatment. Cryptorchidism should be treated early in life, preferably between 6 and 12 months of age or as soon as the diagnosis is established, in order to prevent further damage to spermatogenesis. Recent recommendations strongly favor surgery,8 which offers the best balance between efficacy and side effects. Routine biopsy at surgery is not recommended except in cases of chromosomal abnormality or ambiguous genitalia, where there is an increased risk of gonadoblastoma.
The success rate (defined as a scrotal testis without atrophy) after surgical treatment in expert hands has improved over the years and is now approaching >95% for inguinal testes and 85% to 90% for abdominal testes, with slightly varying results depending on techniques.10 Side effects include pain, hematoma, infection, and anesthetic complications. Complications such as testicular atrophy and damage of the vas deferens are rare but become more frequent if surgery needs to be repeated due to relapse.
Hormonal treatment with human chorionic gonadotropin (hCG), gonadotropin-releasing hormone (GnRH), or luteinizing hormone–releasing hormone (LHRH) are now rarely used; their overall efficacy is only 20%, and reascent appears in approximately 25%.56 The lower the initial testis position, the higher the rate of success. Acute side effects include pain at injection site, penile growth, pubic hair, erection pain, behavioral problems, and pain in the groin. More severely, germ cell apoptosis and inflammatory changes in the testes with the risk of long-term reduction in germ cell number and testis size in adulthood have been reported, especially for the youngest children.57,58 Hormonal treatment after orchidopexy has been proposed to have beneficial effects on sperm count,59 but this treatment regimen needs further scrutiny before it can be generally recommended.
Currently, treatment of retractile testes and high scrotal testes is not recommended because systematic studies are not yet available to assess whether treatment would be of long-term benefit. However, some retractile testes may show histologic deterioration and a reduction in volume over time.60
LONG-TERM EFFECTS
Impairment of spermatogenesis is the major problem of cryptorchidism. This impairment is partly related to a dysgenetic development in utero, but failure or delay to treat will additionally damage future semen quality and fertility.61 The precise adverse mechanisms that affect testicular function in untreated cryptorchid testes are not yet clear.62 Although semen quality is reduced in unilateral cryptorchidism, reported fertility rates are usually not compromised.11,63 In contrast, bilateral cryptorchidism will lead to azoospermia if not treated.64 Long-term follow-up studies show that treatment before 4 years of age improves semen quality outcome. Whether the current recommendation for treatment at an even younger age will lead to further improvement of the long-term outcome remains to be seen. In a prospective study of unilateral cryptorchidism, the affected testis grew better over the first 2 years of life if orchidopexy was performed early.65
Hormonal effects related to cryptorchidism are very subtle in childhood, and controversial results have been reported.66–68 At 3 months of age, gonadotropins and inhibin B levels may already reflect the severity of cryptorchidism, with increasing FSH and decreasing inhibin B levels being found in patients with high scrotal testes with spontaneous postnatal descent, to severely and persistently cryptorchid testes.68 Low serum inhibin B levels at orchidopexy may reflect a low number of germ cells at biopsy.69 In adults, the predominant effect of cryptorchidism is impaired Sertoli cell function, reflected in higher FSH and lower inhibin B levels. The effect on Leydig cell function, reflected by increased luteinizing hormone (LH) and low or normal testosterone level, is quite common but more pronounced70,71 if the orchidopexy was performed at a relatively late age.
Cryptorchidism increases the risk of testicular cancer four- to fivefold,72 and both conditions share several prenatal and perinatal risk factors.7 The relative risk of neoplasia is greatest in the undescended testis (RR = 6.3) but is also slightly increased in the contralateral, normally descended testis in a male with unilateral cryptorchidism.73 Historical data suggest that orchidopexy before the age of 11 years lowered the risk of testis cancer,74 but the outcomes of surgery at even earlier ages require further long-term follow up to determine if testicular cancer rates decline further.
Hypospadias
Hypospadias is a common malformation in newborn boys, but there is still limited knowledge about its etiology. Traditionally, hypospadias is categorized into glanular, coronal, penile, penoscrotal, and perineal hypospadias (Fig. 138-3). Hypospadias may be diagnosed in isolation or in combination with other genital malformations such as cryptorchidism or complex syndromes.
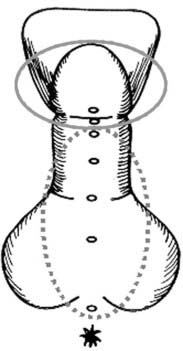
FIGURE 138-3. Schematic presentation of hypospadias. Solid line circle indicates distal hypospadias with urethral opening at the area of glans or corona penis. Broken line circle shows proximal hypospadias, where a cleft opens at the shaft of the penis or in the perineal region. In those cases, the cleft is open all the way to the glans, which is not visible here.
EMBRYOLOGY
After formation of the testis from the undifferentiated gonad, the primitive phallus begins to grow from week 8 in gestation under the influence of fetal androgens, especially dihydrotestosterone (DHT).16 In parallel, the inner genital folds fuse in midline, and the penile urethra forms from the endodermal urethral plate in a proximal-distal direction. Thereafter, the glandular urethra is formed by distal proliferation and canalization of the urethral plate into the glans penis. The preputial skin enlarges from a dorsal fold to eventually cover the ventral surface of the glans and fuse. Hypospadias usually arises in the first trimester as a midline fusion defect of the urethral folds or lack of normal cannulation between weeks 8 and 14 in gestation, leading to a ventral positioning of the meatus and agenesis of the surrounding tissues.
Very mild types of hypospadias may present with a normally shaped prepuce or with isolated chordee and normal placement of the urethral meatus and do not need surgical correction. In rare cases, only a hooded foreskin is found, with a normal urethral opening. In contrast, severe hypospadias with early disturbances of urethral fusion may compromise the growth of the periurethral tissue and thereby lead to chordae and ventral curvature of the penis. In very severe cases, which are often combined with bifid scrotum and cryptorchidism, the clinical appearance of the external genitalia may cause considerable difficulties in gender assignment. Such cases need thorough assessment according to the guidelines for children with disorders of sexual development; there may be additional other malformations of the internal genitalia.75 The true severity of hypospadias may first be established during surgery.
ETIOLOGY
The etiology of hypospadias remains obscure in many cases, and evidence suggests that it is a multifactorial disease with genetic and environmental components. Familial cases account for approximately 10% of all hypospadias, but truly monogenic disorders, such as 5α-reductase gene (SRD5A2) mutations and polymorphisms, MAMLD1 (CXorf6) mutations, Wilms tumor gene 1 (WT1) mutation or chromosomal aberrations are seldom found.76–76b Monozygotic twinning appears to increase the risk of hypospadias by yet unknown mechanisms.77 A recent study described reduced semen quality, increased need for fertility treatment, and a higher rate of genital disorders in fathers of sons with hypospadias.76 Assisted reproduction may contribute to a higher prevalence of hypospadias, potentially through the higher rate of twinning, compromised in utero growth, and paternal subfertility.78 Surveys of large patient groups, however, have not found genetic causes in the majority of cases.79,80
A large number of complex syndromes are associated with hypospadias, including the Smith-Lemli-Opitz syndrome or the hand-foot-genital syndrome.81 Such entities should be kept in mind, especially in cases with concomitant other malformations and/or psychomotor developmental delay. Hormonal disorders with impaired androgen synthesis or action may cause hypospadias, such as inborn errors of steroidogenesis (17β- and 3β-hydroxysteroid dehydrogenase deficiency), partial androgen-receptor insensitivity (Reifenstein syndrome), and 5α-reductase deficiency.82
Hypospadias shares some risk factors with other diseases of the male reproductive tract, in particular low birth weight, being born small for gestational age, and low parity.3,39,83–86 Preeclampsia, placental insufficiency and retention, weak contractions during delivery, increased rate of caesarean sections, and increased need for assisted reproductive techniques due to parental infertility have also been reported in hypospadias. There is a strong association between intrauterine and postnatal growth and hypospadias, although current knowledge does not enable distinguishing the direction of this association. The gender dimorphism of birth weight is at least partly linked to androgen action and is a common adverse factor leading to androgen insufficiency that may compromise both parameters.87,88
Studies on environmental and lifestyle factors in relation to hypospadias are sparse, but exposure to pesticides used in agriculture,40,85 residency close to hazardous waste sites, and vegetarian diets89 appear to influence the risk of hypospadias.90 It is reasonable to assume that particular environmental factors with antiandrogenic effects, such as some fungicides, may disturb the development of the penis and urethra. The use of progestins and diethylstilbestrol have also been linked with hypospadias.43,91 The measurement of AGD in rodents is a biomarker of androgen effect and used in toxicologic research. There is emerging evidence that this measurement may also prove useful in humans. One study reported shorter AGD in boys born after high phthalate exposure in the second trimester of pregnancy.51 Another showed that boys with hypospadias and cryptorchidism had a significantly shorter AGD than controls, indicative of some degree of undervirilization.92
PREVALENCE
The reported prevalence of hypospadias ranges from 2 to 90 per 10,000 male births, depending on geographical region, ethnicity, and study design. Several countries in Europe (Denmark, the Netherlands), the United States, and Western Australia appear to have experienced an increase in the prevalence of this malformation in the last few decades,3,93–97 but others report a stable rate.98 In registries, mild forms such as glanular hypospadias are often underreported.13,99 In addition, mild hypospadias with an intact prepuce may not be recognized at birth because of the high rate of physiologic phimosis.3 However, the reported increase in prevalence was mainly due to an increase in the rate of severe forms. Hypospadias can occur as an isolated form and together with additional malformations of the urogenital tract16 and other internal malformations. The reported relative contribution of mild (glanular) forms to the total prevalence ranges from 29% to 72% and is highly dependent on the age and selection criteria for participation in studies and the completeness of recruitment.13 This causes considerable difficulties in comparing data across countries and over time.
TREATMENT
Very mild types of hypospadias may not require any treatment for either functional or cosmetic reasons, but severe cases will require surgical correction. The aims of surgical treatment are to extend the urethra to the glans penis to allow normal micturition, straightening of the penile shaft to allow normal intercourse, and improvement of cosmetic appearance. As for cryptorchidism, the age of surgical treatment has decreased,100 and there is general consensus that surgery should be done by experts in the field. Cosmetic results with surgery are typically good, but complications such as wound infections, hematoma, urethral fistula, and stricture can cause severe problems.
LONG-TERM EFFECTS
There are only few studies on the impact of unsuccessful surgery on life quality in patients with hypospadias.101 The current debate about the ideal age at correction is unresolved, varying from a recommendation for early surgery100 for cosmetic reasons to awaiting an age at which the patient himself can make an informed decision on treatment options and risks.75
Few studies have focused on the consequences of hypospadias on semen quality, fertility, and sexual function in adulthood. However, there are some studies that report men with hypospadias to have fewer partners, fewer children, a higher need for fertility treatment owing to impaired semen quality, and sexual problems such as genital pain, mechanical difficulties, and ejaculation difficulties.102,103 A recent study showed that only men with severe forms of hypospadias combined with cryptorchidism had poor semen quality, whereas most men with mild hypospadias had normal semen quality, indicating that isolated hypospadias most often is not associated with testicular dysgenesis.103a
Testicular Tumors
It is important that endocrinologists keep in mind a possibility of neoplasia in patients with fertility problems or other reproductive disorders, especially in view that testicular tumors have recently become more common. In the vast majority of cases, the neoplasm is derived from germ cells, and most of these tumors occur in young men. Strong environmental associations have led to germ cell tumors being considered a part of TDS7 (see Fig. 138-1). Somatic cell tumors, known as sex cord–stromal neoplasms, and Leydig cell tumors are much less frequent, have a predominantly genetic background, and are not considered to be a part of TDS. However, regardless of the pathogenesis, nearly all types of overt testicular neoplasms may cause early endocrine effects, and the management nearly always causes some degree of late endocrine complications.
GERM CELL TUMORS
Germ cell tumors account for an estimated 90% to 95% of cases of testicular cancer and are by far the most frequent neoplasms of this organ.104 These tumors have important differences in comparison with somatic solid tumors. The initiation of neoplastic transformation of germ cells begins early in life, and the prevalence of GCT is greatest in young individuals, with the exception of the rare spermatocytic seminoma of elderly men. Secondly, GCTs are characterized by a striking variety of morphologic forms104 (Fig. 138-4). The majority of them are extremely radio- and chemosensitive, except the highly differentiated mature teratomas that are clinically more benign but also less sensitive to treatment. Finally, the incidence of testicular germ cell cancer has been steadily rising in recent decades, in concert with other abnormalities of the male reproductive tract but with marked geographic and ethnic differences.1,5,105
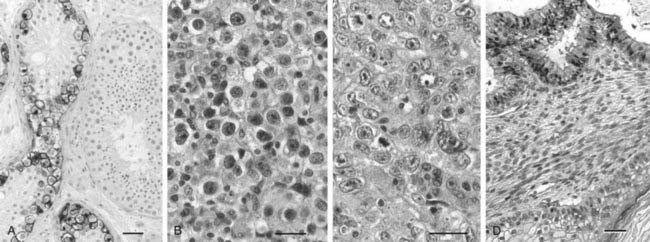
FIGURE 138-4. Histology of the most frequent germ cell neoplasms. All bars 20 µm. A, Preinvasive carcinoma in situ (CIS). CIS cells are stained immunohistochemically with an anti-PLAP (placenta-like alkaline phosphatase) antibody. Note the difference between the CIS-containing tubules and the adjacent larger tubules with normal spermatogenesis. B, Seminoma; tumor cells closely resemble CIS cells. C, Embryonal carcinoma, the undifferentiated component of nonseminomas. D, Teratoma, the most differentiated component of nonseminomas, mimicking embryonic development of somatic tissues.
CLASSIFICATION AND HISTOPATHOLOGY
Because of the heterogeneity of germ cell–derived testicular tumors, several classifications exist,104 of which the one proposed and recently modified by the World Health Organization is most commonly used.106 GCTs occur in three age groups107 (Table 138-1). The first group includes rare germ cell tumors of early childhood (infantile tumors, mainly mature teratoma and yolk sac tumor). The second group of tumors occurs in adolescents and young adults, and because these are by far the most frequent, they are of greatest importance for endocrinologists. These tumors include seminoma, nonseminomatous tumors, and combined tumors. The combined (mixed) tumors contain elements of seminoma and nonseminomatous tumors but clinically are treated as nonseminoma. Finally, in older age, spermatocytic seminoma is a predominant type of tumor. A characteristic feature of the tumors of young adults is that they originate from a preinvasive lesion, carcinoma in situ (CIS) testis,108,109 also known as the intratubular germ cell neoplasia (ITGCN) or testicular intraepithelial neoplasia (TIN) (see Fig. 138-4). The infantile tumors and spermatocytic seminoma are not preceded by CIS and apparently have a different pathogenesis.107,109
Table 138-1. Testicular Germ Cell Tumors
ITCN, Intratubular germ cell neoplasia; TIN, testicular intraepithelial neoplasia.
Another preinvasive lesion closely related to CIS is gonadoblastoma, which occurs almost exclusively in individuals with disorders of sexual differentiation (DSD), both with predominantly female and male phenotype, most frequently though in patients with mixed gonadal dysgenesis, 45,X/46,XY.110 Gonadoblastoma consists of nests of gonocyte- or CIS-like cells surrounded by stromal cells resembling primitive granulosa cells.111,112 The affected gonad in most cases is abnormal and may contain testicular cordlike structures (sometimes containing CIS cells), ovarian-like elements, or atrophic streak gonad tissues in various proportions.110,113 The presence of gonadoblastoma or CIS depends on the relative degree of virilization of Sertoli cells surrounding the germ cells; the less virilized the cells are, the more likely is the presence of gonadoblastoma-like structures. A good marker of masculinization is Sertoli cells is SOX9, which is present in tubules with CIS cells, whereas somatic granulosa-like cells surrounding gonadoblastoma cells express primarily FOXL2.113a The clinical course is usually benign; however, gonadoblastoma can in rare cases transform into a malignant GCT. No particular endocrine manifestations of gonadoblastoma have been noted other than the endocrine problems inherent to a background problem with sex differentiation and the type of DSD.
As far as morphology is concerned, both CIS and gonadoblastoma cells resemble gonocytes, or immature germ cells. CIS cells are located inside seminiferous tubules in the place normally occupied by spermatogonia (see Fig. 138-4), while gonadoblastoma cells form clusters of cells intertwined with granulosa-resembling somatic cells and do not retain a tubular structure. More subtle dysgenetic features, such as undifferentiated Sertoli cells or the presence of intratubular hyaline bodies (also visible as microlithiasis in the scrotal ultrasound) are frequently seen in testes with CIS and overt tumors (Fig. 138-5), as well as in cryptorchid testes (even without neoplasia), consistent with the TDS hypothesis.7,114,115 CIS cells (and most likely also gonadoblastoma cells) share the gene expression profile with a subset of fetal and early infantile germ cells18,116 and to some extent with embryonic stem cells,117 thus the current hypothesis stipulates that CIS cells are transformed gonocytes.118,119
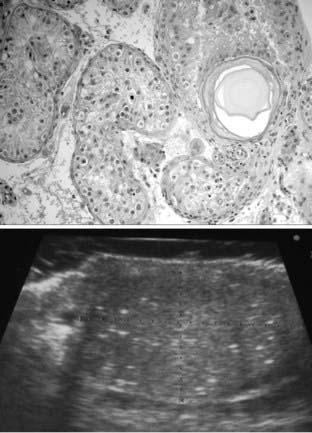
FIGURE 138-5. Signs of testicular dysgenesis revealed by testicular biopsy or scrotal ultrasound examination. Top, tubules with carcinoma in situ (CIS) cells and a large microlith in a biopsy of a young adult man. Bottom, ultrasound image of testicular microlithiasis.
Seminoma cells resemble CIS cells and grow as a homogeneous tumor that retains features of germ cells (see Fig. 138-4). Seminomas are diagnosed in the 25- to 40-year-old age group, whereas nonseminomatous tumors occur in relatively younger men (17 to 30 years) and usually have a more severe clinical course. Nonseminomas display a variety of histologic forms, ranging from undifferentiated embryonal carcinoma to mature teratoma104 (see Fig. 138-4). Among nonseminomatous components, two extraembryonic types deserve special note because of their endocrine activity: choriocarcinoma, which resembles gestational trophoblast and produces large quantities of hCG, and yolk sac tumor, which is similar in morphology to the embryonic yolk sac and secretes alpha fetoprotein (AFP).120,121
EPIDEMIOLOGY AND RISK GROUPS
The average age-adjusted incidence of testicular GCT has been markedly increasing among whites, in particular in northwest Europe and North America, thus this cancer became the most frequent malignancy among young men in these regions.1,5 Epidemiologic studies documented that cryptorchidism is a strongly predisposing condition for testicular cancer and revealed an almost identical pattern of risk factors, consistent with a pathogenetic link between the two diseases.55,72,122 Among risk factors for testicular cancer, the following are most frequently reported: low birth weight,123 premature birth, birth order,124 high levels of maternal estrogens or bleeding during pregnancy,125 tall stature,126 high maternal age or body weight of the mother, and neonatal jaundice. Nearly all these risk factors are operating pre- or perinatally.7,118,127,128
Individuals with developmental abnormalities of the gonads and DSD are at high risk of germ cell neoplasia. Conditions with an abnormal karyotype, especially those involving aneuploidy of sex chromosomes and structural abnormalities of chromosome Y129–131 but also Down syndrome,132 carry a high risk of germ cell neoplasia. Individuals with the androgen insensitivity syndrome, whose gonocytes differentiate to spermatogonia with a substantial delay, are also at risk of CIS testis,133 but those with the complete form of this syndrome rarely develop overt tumors.134,135
Patients with a unilateral testicular GCT are at increased risk for the CIS testis and development of a testicular tumor in the contralateral testis, especially in a testis with the presence of testicular atrophy or impaired spermatogenesis.136,137 In fact, the first cases of testicular CIS were described in patients referred for infertility,108 and infertility is clearly a risk factor for CIS and testicular cancer.138,139 Sperm production in patients with testicular cancer is often markedly lower than controls, and these patients have significantly decreased offspring sex ratio and lower fertility rates, even prior to development of their tumor.137,140–143
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


