FIGURE 94-1. Transport, deiodination, and nuclear action of thyroid hormones. Transporters are required for passage of T3 and T4 across the plasma membrane, facilitating hormone uptake, efflux, or both. Deiodinases catalyze conversion of T4 to T3 (D1, D2) or inactivation of T4 to rT3 and T3 to T2 (D3). T3 interaction with its nuclear receptor (TR), usually part of a heterodimer with RXR, modulates target gene transcription and protein synthesis.
Differential Diagnosis of Elevated T4, T3 With Nonsuppressed TSH
A number of genetic disorders and clinical contexts are associated with elevated thyroid hormones and nonsuppressed thyroid-stimulating hormone (TSH) levels (Table 94-1). The first step in making a diagnosis is to verify the validity of hormone measurements. Confirmation of elevated free thyroid hormone levels in two-step or equilibrium dialysis assays excludes abnormal circulating binding proteins or antiiodothyronine antibodies. Preservation of linearity when TSH is assayed in dilution suggests this measurement is not artifactual. Many causes (nonthyroidal illness, psychiatric disorder, neonatal period, drugs) can be excluded by clinical context.
Table 94-1. Causes of Elevated Thyroid Hormones With Detectable TSH
Genetic disorders associated with elevated thyroid hormone levels can also be distinguished on the basis of different patterns of abnormal thyroid function (Table 94-2). The basis for such distinct biochemical profiles in each disorder is described later.
Resistance to Thyroid Hormone
CLINICAL FEATURES
The syndrome of resistance to thyroid hormone (RTH) is characterized by elevated circulating levels of free thyroid hormones together with nonsuppressed pituitary TSH secretion. It reflects resistance to thyroid hormone action in the hypothalamic-pituitary-thyroid (HPT) axis and is accompanied by variable refractoriness in peripheral tissues.
RTH was first described in 1967 in two siblings who were clinically euthyroid despite high circulating thyroid hormone levels. The siblings exhibited several other abnormalities, including deaf-mutism, stippled femoral epiphyses with delayed bone maturation and short stature, and dysmorphic facies, winging of the scapulae, and pectus carinatum.3 It is now clear that some of these features are unique to this kindred in whom the disorder was recessively inherited. The majority of RTH cases that have been subsequently described are dominantly inherited, with a highly variable clinical phenotype. Affected subjects are either asymptomatic or have nonspecific symptoms and may be noted to have a goiter, prompting thyroid function tests that suggest the diagnosis. In these individuals, classified as exhibiting generalized resistance to thyroid hormone (GRTH), the high thyroid hormone levels are thought to compensate for ubiquitous tissue resistance, resulting in a euthyroid state. In contrast, a smaller number of individuals (around 15%) who share the same biochemical phenotype exhibit clinical features of thyrotoxicosis. In adults, these can include weight loss, tremor, palpitations, insomnia, and heat intolerance; in children, failure to thrive, accelerated growth, and hyperkinetic behavior have also been noted. When this clinical entity was first described, patients were thought to exhibit “selective” pituitary resistance to thyroid hormone (PRTH) action, with preservation of normal hormonal responses in peripheral tissues,4 but it is now recognized that peripheral resistance (typically hepatic) to hormone action is present even in these subjects. Less commonly, hypothyroid features such as growth retardation, delayed dentition, and bone age in children or asthenia and hypercholesterolemia in adults have been observed in RTH and may even coexist with thyrotoxic symptoms in the same individual.5 Taken together, these observations suggest that the clinical features of this disorder are influenced by the degree of refractoriness of peripheral tissues to high circulating levels of free thyroid hormones.
The estimated prevalence of RTH is 1 in 50,000 live births; the disorder can be diagnosed neonatally by screening with a combination of TSH and free T4 measurements.6 Over 700 cases of RTH (from more than 250 families) have now been described worldwide, enabling clinical characteristics of this disorder to be defined more precisely.
Goiter
A palpable goiter has been documented in 65% of individuals, particularly adult females. The enlargement is usually diffuse, with multinodular glands being typical of recurrent goiters following partial thyroidectomy. Development of toxic multinodular goiter on the background of RTH has been documented in a single case.7 Interestingly, it has been noted that fewer children with RTH born to affected mothers exhibit thyroid enlargement (35%) compared to offspring of unaffected mothers (87%), suggesting that maternal hyperthyroxinemia with transplacental passage of thyroid hormones during development might protect against goitrogenesis.8 The bioactivity of circulating TSH has been shown to be significantly enhanced in RTH, perhaps accounting for the goiter and markedly elevated serum thyroid hormones, despite the normal immunoreactive TSH levels observed in many cases.9
Cardiovascular System
Palpitations and resting tachycardia have been reported in approximately 75% of those with GRTH and almost all cases of PRTH, with a particular predisposition to atrial fibrillation in older subjects.8 The incidence of these symptoms is notably higher in RTH patients than in unaffected relatives or in the general population, although still less frequent when compared to patients with classic hyperthyroidism.10 In one study, although resting heart rates were comparable to unaffected family members, 30% of RTH subjects showed echocardiographic features of increased myocardial contractility and impaired diastolic relaxation, with a greater incidence of mitral valve prolapse.8 In a prospective study of cardiovascular involvement in a large cohort of children and adults with RTH, resting heart rate was significantly higher. Some indices of cardiac systolic and diastolic function (e.g., stroke volume, cardiac output, maximal aortic flow velocity) were intermediate between values in normal and hyperthyroid subjects. Other parameters (e.g., ejection and shortening fractions of the left ventricle, systolic diameter, and left ventricle wall thickness) were not different, indicating a partially hyperthyroid response of the heart in this disorder.10 Systemic vascular resistance and arterial stiffness are increased in RTH.11,12
Musculoskeletal System
Stippled epiphyses and winged scapulae were noted in the original RTH kindred but have not been observed in other cases. These features may represent a specific manifestation of the known gene deletion (TRβ) or an unrelated genetic abnormality in this consanguineous kindred.3 In contrast, growth retardation and delayed skeletal maturation are more common in childhood RTH patients, with height below the fifth percentile in 18% and delayed bone age (>2 SD) in 29%,8 with no significant differences between GRTH and PRTH cases. Despite these abnormalities, final adult height is often unaffected.13
Bearing in mind the known adverse effects of untreated hyperthyroidism on bone mineralization, we have conducted a cross-sectional survey of approximately 80 adult subjects with RTH and observed a reduction in bone mineral density in the femoral neck (mean Z score −0.71) and lumbar spine (mean Z score −0.73) but with normal markers of bone turnover (Chatterjee and Beck-Peccoz, unpublished observations).
Basal Metabolic Rate
The basal metabolic rate (BMR) is variably affected in RTH, being normal in some cases.14 In keeping with others,8 we have observed an elevated BMR, particularly in childhood RTH (Gurnell, Chatterjee, and Beck-Peccoz, unpublished observation), which may account for the abnormally low body mass index seen in approximately a third of children.
Central Nervous System
Two studies have documented neuropsychological abnormalities in RTH. First, a history of attention deficit hyperactivity disorder (ADHD) in childhood was elicited more frequently in patients with RTH (75%) compared to their unaffected relatives (15%).15 A second study showed that both children and adults with RTH exhibited problems with language development, manifested by poor reading skills and problems with articulation (e.g., speech delay, stuttering).16 Frank mental retardation (IQ < 60) is relatively uncommon (3%), although 30% of patients show mild learning disability (IQ < 85), probably due to uncompensated CNS hypothyroidism.14 A direct comparison of individuals with ADHD and RTH versus ADHD alone indicates an association with lower nonverbal intelligence and academic achievement in the former group.17 In detailed analysis of one family, RTH cosegregated with lower IQ rather than ADHD,18 so it is possible that low IQ facilitates the manifestation of ADHD. However, two different surveys of unselected children with ADHD failed to detect any cases of RTH by biochemical screening, suggesting that the latter disorder is unlikely to be a common cause of hyperactivity.19,20 Although magnetic resonance imaging (MRI) shows that anomalies of the sylvian fissure or Heschl’s gyri are more frequent in RTH, these features do not correlate with ADHD.21
Hearing and Vision
Significant hearing loss has been documented in 21% of RTH cases, similar to the prevalence reported in congenital hypothyroidism.8 In the majority, audiometric tests indicated a conductive defect, probably related to the increased incidence of recurrent ear infections in childhood RTH (67% in RTH versus 28% in normal controls). Abnormal otoacoustic emissions, consistent with cochlear dysfunction, have also been documented in those with hearing deficit,8,22 and cochlear expression of TRβ has been shown.23 The single kindred with deaf-mutism and recessively inherited RTH harbored a complete deletion of the TRβ gene,3 which correlates with the finding that TRβ knockout (KO) mice are deaf.24,25 Together, these observations underscore the importance of TRβ in auditory development and function. Deletion of the TRβ2 isoform in mice is associated with selective loss of M-cone photoreceptors and abnormal color vision,26 but monochromatic color vision has only been reported in the rare human kindred with recessively inherited RTH and a complete TRβ gene deletion.3 Detailed assessment of 10 subjects with TRβ point mutations and dominantly inherited RTH showed no common color-vision abnormalities (Gurnell and Chatterjee, unpublished observations).
Other Associated Disorders
Rarely, cases of RTH have been described where coexistent autoimmune thyroid disease has also been documented,27–29 raising the possibility of a pathogenic association between these disorders. Coexistence of Pendred syndrome and RTH has been documented in a single case.30 Pituitary enlargement, as a consequence of impaired negative-feedback regulation of TSH secretion, is another potential association with RTH. While pituitary hyperplasia has been reported in a single case, it occurred in the context of massively elevated TSH levels with suboptimal thyroxine replacement therapy following inappropriate thyroid ablation and regressed once TSH levels normalized.31 Only a few cases of RTH associated with pituitary adenomas have been described,32,33 suggesting that pituitary hyperplasia or adenoma formation are uncommon clinical sequelae in RTH, provided the altered set-point of the HPT axis is not perturbed. A greater frequency of recurrent upper respiratory tract and pulmonary infections has been reported in RTH, and affected individuals have reduced serum immunoglobulin levels.8 A retrospective study of a large Azorean kindred has shown a higher rate of miscarriage in mothers affected by RTH, with unaffected offspring being of lower birth weight, suggesting that intrauterine exposure to high TH levels does have adverse fetal effects.34
Differential Diagnosis
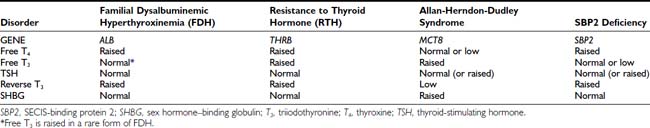
Differentiation of RTH, particularly the form associated with hyperthyroid features from a TSH-secreting pituitary tumor, can be difficult (Fig. 94-2). Similar abnormalities in thyroid function tests in first-degree relatives strongly suggest RTH, together with normal pituitary imaging and serum α-glycoprotein subunit levels. Other factors that are helpful in making this differential diagnosis, such as dynamic testing and clinical/biochemical features, are discussed in greater detail in Chapter 17 (TSH-Producing Adenomas).
The development of Graves disease in RTH is suggested by atypical features such as ophthalmopathy, severe thyrotoxic symptoms, and a further rise in thyroid hormones leading to a subnormal or suppressed TSH. Following antithyroid drug treatment, TSH levels become elevated despite normalization of thyroid hormones. Similarly, normalization of TSH but elevated thyroid hormones following supraphysiologic doses of thyroxine replacement in primary hypothyroidism suggests coexistent RTH.
In addition to clinical features, the measurement of various tissue markers of thyroid hormone action has been suggested to be a useful method for evaluating the differing responses of various target organs and tissues to elevated circulating thyroid hormones (Table 94-3). These measurements are most useful in assessing the tissue effects of marked thyroid hormone excess (as typically found in overt thyrotoxicosis) but may be less discriminatory in borderline hyperthyroidism or in hypothyroidism. To improve the sensitivity and specificity of these parameters, it has been suggested that individuals with RTH be assessed by measuring tissue responses dynamically following the administration of graded supraphysiologic doses of T3 (50, 100, and 200 µg/day, each given for a period of 3 days), with comparison of any change in indices from baseline values to those observed in normal subjects.35
Table 94-3 Tissue Indices of Thyroid Hormone Action
| Pituitary: | Thyroid-stimulating hormone (TSH) |
| General: | Basal metabolic rate (BMR) |
| Hepatic: | Sex hormone–binding globulin (SHBG), ferritin, cholesterol |
| Muscle: | Creatine kinase, ankle jerk relaxation time |
| Bone: | Height, bone age, bone density, osteocalcin, alkaline phosphatase, pyridinium crosslinks, type 1 collagen telopeptide |
| Cardiac: | Sleeping pulse rate, systolic time interval, diastolic isovolumic relaxation time |
| Hematologic: | Soluble interleukin 2 receptor (sIL-2R) |
| Lung: | Angiotensin-converting enzyme (ACE) |
MOLECULAR GENETICS
TRβ RTH
Following the cloning of TRα and TRβ, RTH was shown to be tightly linked to the TRβ gene locus in a single family.36 This prompted analysis of the TRβ gene in other cases, and a large number of receptor defects have since been associated with this disorder. Eighty percent of RTH is familial, dominantly inherited, and associated with heterozygous mutations in the TRβ gene14,37–39; de novo receptor mutations occur in the remaining 20% of sporadic cases. Over 100 different defects including point mutations, in-frame deletions, and frameshift insertions have been documented to date, which localize to three mutation clusters within the ligand-binding domain of the receptor (Fig. 94-3). Within each cluster, some codon changes (e.g., R243W, R338W, R438H) representing transitions in mutation-prone CpG dinucleotides occur more frequently and are overrepresented.40
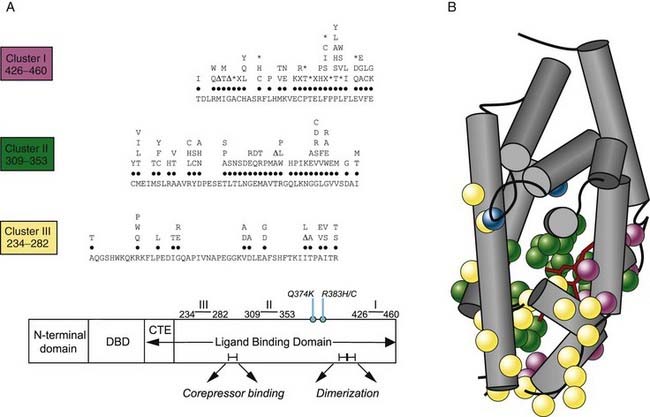
FIGURE 94-3. A, A schematic representation of the domains of TR-β showing that with two exceptions (Q374K, R383H/C), RTH receptor mutations localize to three clusters within the ligand-binding domain (LBD). The receptor defects described include different missense substitutions at each codon, in-frame codon deletions (Δ), premature termination codons (X), and frameshift mutations (*). The mutations shown include those listed in a public database (HGMD) together with our unpublished data. No RTH receptor mutations have been described in the zinc-finger DNA-binding domain (DBD) or its carboxy-terminal extension (CTE), which together mediate interaction with DNA, or regions in the LBD important for corepressor binding or dimerization with RXR. B, The crystal structure of the TR-β ligand-binding domain (LBD) (Protein Data Bank accession no. 1BSX) composed of 12 α helices (gray) is shown, with the location of missense mutations associated with RTH (cluster I, purple; cluster II, green; cluster III, yellow; R383,Q374, blue) superimposed. As predicted from their functional properties, the majority of mutations involve residues surrounding the ligand-binding (T3, red) cavity.
Based on the supposition that PRTH was associated with selective pituitary resistance, it had been hypothesized that this disorder might be associated with defects in DIO2 or the TRβ2 receptor isoform, but a number of reports have documented TRβ mutations in PRTH.38,41,42 Receptor mutations found in individuals with PRTH have also been associated with GRTH in unrelated kindreds. Furthermore, even within a single family, the same receptor mutation can be associated with abnormal thyroid function and thyrotoxic features consistent with PRTH in some individuals but similar biochemical abnormalities and a lack of symptoms indicative of GRTH in other members. Overall, these findings indicate that GRTH and PRTH represent the phenotypic spectrum associated with a single genetic entity.
Non-TRβ RTH
In a small but significant number of cases (10% to 15%), clear-cut biochemical evidence of RTH is not associated with a mutation in the coding region of TRβ—so-called non-TRβ RTH. One explanation for this is somatic mosaicism, with occurrence of a TRβ mutation whose expression is selective, being detectable in some tissues but not peripheral blood leukocyte DNA.43 Alternatively, defects in other proteins involved in TR signaling have also been postulated. This latter hypothesis is supported by the description of kindreds with thyroid function tests and resistance to exogenous T3 similar to subjects with TRβ RTH, but in whom linkage and sequence analyses have excluded defects in TRβ and TRα genes.44,45 While it is theoretically possible that defects at any point in the pathway of thyroid hormone action could manifest as an RTH phenotype, evidence exists to favor some candidate genes such as RXR or the cofactors (e.g., corepressors, coactivators, TR-associated proteins) that regulate thyroid hormone–dependent gene transcription.
Mice harboring a deletion of the SRC-1 gene show abnormalities in thyroid function tests suggestive of RTH, together with subtle evidence of resistance in other steroid receptor axes.46 Similar findings were noted in mice doubly heterozygous for knockouts of the SRC-1 and transcriptional intermediary factor 2 (TIF-2) coactivator genes.47 To date, no homologous human disorder has been described, with linkage studies and direct sequence analysis of several cofactor genes (e.g., SRC-1, SRC-3, SMRT) in non-TRβ RTH kindreds or individuals failing to identify any abnormality.49 In one case, wild-type TRβ was found to exhibit aberrant binding to a unique 84-kD protein from patient but not control fibroblast nuclear extracts, suggesting abnormal receptor interaction with a cofactor44 whose identity has not been elucidated. It is known that patients with Rubinstein-Taybi syndrome, a disorder associated with heterozygous defects in the nuclear receptor coactivator CBP, exhibit a number of somatic abnormalities (broad thumbs, mental retardation, short stature) yet have normal circulating free T4 and TSH levels,50 indicating that mutations in this cofactor are not a cause of non-TRβ RTH. Several lines of evidence favor RXR as a candidate gene in non-TRβ RTH. First, knockout mice lacking the RXRγ isoform, whose tissue expression is limited but includes pituitary thyrotrophs, exhibit thyroid hormone resistance together with an increased metabolic rate.51 Second, the administration of RXR-selective agonists in humans inhibits pituitary TSH secretion, resulting in central hypothyroidism.52 Finally, in two kindreds with non-TRβ RTH, possible linkage to the RXRγ gene locus was noted,45,48 but in another study, no RXRγ gene mutations were identified in four non-TRβ RTH subjects.53 Together, these observations suggest that defects in pituitary-expressed RXRγ might also impair negative feedback in the pituitary-thyroid axis and manifest as RTH. Finally, it is tempting to speculate that a combination of “less functionally deleterious” mutations or even polymorphisms in several genes involved in thyroid hormone action could result in an RTH phenotype, representing an oligogenic basis for the disorder.
PROPERTIES OF MUTANT RECEPTORS
Consonant with their location in the hormone-binding domain, the majority of receptor mutants identified in RTH exhibit moderate or markedly reduced T3 binding; consequently, their ability to activate or repress target gene expression is impaired.54,55 A subset of RTH mutations associated with markedly abnormal thyroid function in vivo and altered transcriptional function in vitro (but little impairment in ligand binding) have been described. Such natural mutations involve residues that mediate receptor interaction with transcriptional coactivators.39,56 In the first RTH family described, with the recessively inherited form of the disorder, the two affected siblings were found to be homozygous for a complete deletion of both alleles of the TRβ receptor gene.57 Importantly, the obligate heterozygotes in this family harboring a deletion of one TRβ allele were completely normal with no evidence of thyroid dysfunction. This suggested that simple deficiency of a functional β receptor, as a consequence of the single deleted TRβ allele, was insufficient to generate the resistance phenotype. This led to the hypothesis that the heterozygous mutant receptors in dominantly inherited RTH were not simply functionally impaired but also capable of inhibiting wild-type receptor action. Studies confirmed that when coexpressed, the mutant proteins are able to inhibit the function of their wild-type counterparts in a dominant-negative manner.58,59 Further clinical and genetic evidence supporting this notion have been provided by two rare examples of RTH. In the first, a childhood case, severe resistance with marked developmental delay and growth retardation associated with cardiac hyperthyroidism was ultimately fatal due to heart failure following septicemia; this individual was homozygous for a mutation (Δ337T) in both alleles of the TRβ gene.60 In the second more recently reported case, the affected subject also exhibited a particularly severe clinical phenotype and was found to be either homozygous or hemizygous for a TRβ mutation (I280S).61 Presumably, the extreme phenotype observed in both cases reflected not only the absence of normally functioning TRβ but the added dominant-negative inhibitory effect of mutant β receptors.
Functional studies of mutant receptors indicate that although they are transcriptionally impaired and dominant-negative inhibitors, their ability to bind DNA and form heterodimers with RXR is preserved.54,55 Conversely, it has been shown that the introduction of additional artificial mutations that abolish DNA binding or heterodimer formation abrogates the dominant-negative activity of mutant receptors in vitro.55,62,63 Mice heterozygous for a TRβ mutation lacking DNA binding do not exhibit RTH.64 It has also been suggested that the ability of mutant receptors in RTH to repress or “silence” basal gene transcription is likely to be an important factor contributing to their dominant-negative potency. Non-T3-binding mutants exhibit constitutive silencing function, particularly when bound to DNA as homodimers, which cannot be relieved by ligand. Conversely, RTH mutants with impaired homodimerization properties are weaker dominant-negative inhibitors.65 With the identification of corepressors, these observations have been extended to show that some RTH mutants either bind corepressor more avidly when unliganded or fail to dissociate fully from corepressor upon T3 binding.66 Furthermore, artificial mutations that abolish corepressor binding abrogate the dominant-negative activity of RTH receptor mutants.66 It has also been suggested that corepressors mediate basal activation of negatively regulated gene promoters (e.g., thyrotropin-releasing hormone [TRH], TSH-α, TSH-β) by unliganded TR.67 An unusual RTH receptor mutant (R383H) exhibits both delayed T3-dependent corepressor release and impaired hormone-dependent negative transcriptional regulation.68 Given the pivotal role of negatively regulated target genes in the pathogenesis of RTH, aberrant corepressor recruitment or release may well prove to be the critical receptor abnormality in this disorder.
Together, these observations allow a model to be constructed (Fig. 94-4) in which occupancy of target gene binding sites by mutant receptor-corepressor complexes mediates dominant-negative inhibition by RTH mutants. Mapping of the three clusters of RTH receptor mutations identified hitherto on the crystal structure of the ligand-binding domain of TRβ69 provides insights into structure-function relationships in TR (see Fig. 94-3). As expected from their impaired ligand-binding properties, most mutations are located around the hormone-binding cavity, and receptor regions mediating DNA binding, dimerization, and corepressor interaction are devoid of naturally occurring mutations, possibly because they lack dominant-negative activity and therefore elude discovery—being biochemically and clinically silent.
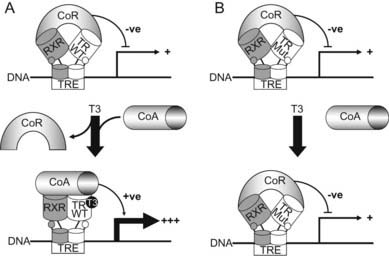
FIGURE 94-4. A model for dominant-negative inhibition by mutant receptors in RTH. Left panel (A)) depicts current understanding of wild-type TR action on target genes. The unliganded TR-RXR heterodimer or homodimer (not shown) recruits a corepressor (CoR) complex to inhibit or silence basal gene transcription. Receptor occupancy by T3 promotes corepressor dissociation and derepression, followed by binding of coactivators (CoA), which leads to target gene activation. Right panel (B) shows RTH mutant receptor action. In comparison to wild-type TR, the primary defect in mutant receptors may be impaired hormone-dependent corepressor dissociation and coactivator recruitment. For the majority of receptor mutants, this functional alteration is a consequence of their reduced ability to bind ligand, but a subset of mutants exhibit enhanced corepressor binding, delayed corepressor release, or impaired coactivator recruitment, with relative preservation of hormone binding. Mutant receptor-CoR complexes compete with their wild-type counterparts for occupancy of promoter thyroid response elements (TREs), resulting in inhibition of target gene expression. In this model, DNA binding, dimerization, and corepressor interaction are functional properties preserved in mutant receptors and required for their dominant-negative activity.
PATHOGENESIS OF VARIABLE RESISTANCE
Genetic and functional evidence suggests that the ability to exert a dominant-negative effect on target genes within the HPT axis is a fundamental property of RTH receptor mutants, generating the abnormal thyroid function characteristic of the disorder. Indeed, some studies indicate that for a subset of RTH mutants, there is a correlation between their functional impairment in vitro and the degree of central pituitary resistance, as measured by the magnitude of elevation in serum-free T4 in vivo.70,71 On this biochemical background, the heterogeneous clinical phenotype may be due to differing degrees of peripheral resistance in different individuals as well as variable resistance in different tissues within a single subject. A number of factors may contribute to such variable tissue resistance.
One important contributory element may be the differing tissue distributions of receptor isoforms. The hypothalamus/pituitary, and liver express predominantly TRβ2 and TRβ1 receptors, respectively, and TRα1 is the major species detected in myocardium. Mutations in the TRβ gene are likely to be associated with pituitary and liver resistance, as exemplified by nonsuppressed TSH and normal sex hormone–binding globulin (SHBG) levels seen in patients, whereas the tachycardia and cardiac hyperthyroidism in some cases may represent retention of myocardial sensitivity to high circulating thyroid hormones acting via a normal α receptor. Another factor that may influence the degree of tissue resistance is the relative expression of mutant versus wild-type TRβ alleles. One study has suggested that both alleles are equally expressed72; another showed marked differences in the relative levels of wild-type and mutant receptor messenger RNA in skin fibroblasts from two RTH cases.73 In one of these individuals, a temporal variation in expression of the mutant allele in fibroblasts appeared to correlate with the degree of skeletal tissue resistance. The dominant-negative inhibitory potency of mutant receptors has been shown to differ depending on target gene promoter context55,74 and is a further variable that may influence the degree of resistance. Finally, factors unrelated to the TRβ gene might influence the phenotype. For example, a deleterious R316H mutation was associated with normal thyroid hormone levels in some members of one kindred75 but clearly abnormal thyroid function in an unrelated family,38 suggesting that other genetic variables can modulate the effect of receptor mutations.
While the absence or presence of overt thyrotoxic features allows patients to be classified as either GRTH or PRTH—a clinical definition that will probably remain useful as a guide to the most appropriate form of treatment—studies indicate that there is some overlap of features between the two forms of the disorder. For example, there are no significant differences in age, sex ratio, frequency of goiter, thyroid function, or clinical features between patients with GRTH or PRTH.76 Importantly, features such as tachycardia, hyperkinetic behavior, and emotional disturbance have been documented in individuals with GRTH.76 Conversely, serum SHBG, a hepatic index of thyroid hormone action, is almost invariably normal in patients with PRTH, suggesting that tissue resistance is not solely confined to the HPT axis in this group of patients.77
Attempts to correlate the phenotype of RTH with the nature of the underlying TRβ mutation have been confounded by three factors: (1) the relative imprecision of clinical criteria used to define GRTH and PRTH; (2) the apparent temporal variation in hyperthyroid features in some RTH cases, such that thyrotoxic symptoms and signs can develop and disappear spontaneously when individuals are followed over several years76; and (3) the relatively small number of patients with any given mutation that have been identified. Nevertheless, some interesting correlations have emerged from the published literature. The first patient reported to have PRTH4 was found to harbor an R338W receptor mutation,41 and the same phenotype has been described in the majority of individuals with this or similar substitutions at this codon.38,42 Interestingly, when tested in vitro, this mutant exhibits dominant-negative activity with the negatively regulated pituitary TSH α subunit gene promoter, but it is a relatively poor inhibitor of wild-type receptor action in other promoter contexts.55 When introduced into other RTH receptor–mutant backgrounds, this mutation weakens their dominant-negative potency on positively regulated reporter genes.78 A patient with the R383H receptor defect, which is impaired mainly in regulation of TRH and TSH genes, exhibited predominant central resistance following T3 administration,79 and the R429Q mutation, with similar functional properties, may also occur more frequently in association with PRTH. Some receptor mutants (R338W or L, V349M, R429Q, I431T) associated with PRTH are either more deleterious80 or exert a greater dominant-negative inhibitory effect in a TRβ2 than a TRβ1 context.81A receptor mutation that selectively fails to bind NCoR but not SMRT is associated with PRTH.82
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


