FIGURE 23-1. Schematic representation of the human GH1 gene showing the alternative splicing of exon 3. This gene contains five exons and four introns; exons are numbered E1 through E5 and introns are shown as A through D. Lengths of these introns and exons are shown in kilobases. As is shown on the left-hand side, normal gene transcription and precursor RNA splicing produces a messenger RNA that is translated to produce a growth hormone (GH) precursor of 217 amino acids (isoform 1). The mature protein, as a result of post-translational cleavage of the signal peptide (which allows it to exit the cell), contains 191 amino acids and has a molecular weight of 22 kDa. As is shown on the right-hand side, alternative precursor RNA splicing gives rise to a variant GH of 20 kDa (isoform 2) that lacks 15 amino acids from the beginning of exon 3 (amino acid residues 58 through 72). This alternative splicing reaction (indicated by the dashed line) takes place because of the presence of a 3′ cryptic alternative splice acceptor site in exon 3. Each GH isoform can undergo further post-translational modification. Several such variants of human (h)GH have been described, including variable phosphorylation of residues Ser132 (white circles) and Ser176 (white circles with black dots). mRNA, Messenger RNA; UTR, untranslated region; P, phosphate group.
(From Kopchick JJ, Sackmann-Sala L, Ding J: Primer: molecular tools used for the understanding of endocrinology, Nat Clin Pract Endocrinol Metab 3[4]:355–368, 2007.)
A family of genes that encodes several transcription factors, including POU1F1 (POU domain, class 1, transcription factor 1, formerly called PIT1) and PROP1 (prophet of PIT1), have been identified and cloned, and have been found to have a major influence on the development of GH-producing cells. Expression of these genes is important in differentiation of pituitary cell lines to somatotrophs that ultimately synthesize and release GH.8 Expression and secretion of GH by somatotrophs are controlled by nutrition, sleep, exercise, and several hormones, as well as by hypothalamic peptides such as GHRH and SS, and GH secretagogues, including ghrelin. This topic is thoroughly covered in this chapter.
GH ACTIVITIES
As the name implies, the major function of GH is growth promotion, regardless of the heterogeneity of GH genes and isoforms and variants of the respective hormones. However, several non–growth-promoting GH actions have been demonstrated, most of which relate to the role of GH in metabolism. We will briefly mention some of these activities here; they are reviewed further in the chapters that focus on the treatment of patients with growth disorders.
Hyposecretion of GH during childhood and adolescence leads to a GH-deficient state associated with dwarfism, whereas hypersecretion of GH before the end of puberty leads to gigantism. These disorders are due to the lack or excess of the growth-promoting action of GH on the bone growth plate. In contrast, during adulthood, when linear growth has already been completed, GH deficiency does not affect growth; however, it does affect body composition, carbohydrate and lipid metabolism, cardiovascular risk profile, and quality of life.9,10 Hypersecretion of GH in adults, mainly derived from pituitary adenomas, results in a clinical condition known as acromegaly that is characterized by soft tissue enlargement, most of which occurs in the acral regions, and involves abnormal growth of several organs, including the heart, liver, and kidneys. Together, these pathologic changes lead to life-threatening conditions, including diabetes mellitus, cardiovascular disease, and sleep apnea.11
In healthy adults, GH displays several metabolic effects, including those noted on protein and fat, although its major effects are exerted on carbohydrate metabolism. Insulin is the main hormone that controls substrate metabolism during the fed state; however, during fasting, when insulin secretion is suppressed, this function shifts to GH.12 Nevertheless, the specific impact of GH on carbohydrate metabolism is not fully understood, with two contradictory actions described: acute or early insulin-like activities, and chronic or late anti-insulin effects. The chronic effect is also described as the diabetogenic activity of GH; acute insulin-like activities include hypoglycemia and increased glucose and amino acid transport and metabolism, with increased protein synthesis,13 increased glycogenesis, and increased lipogenesis.10 These insulin-like activities are seen primarily in vitro or under special in vivo circumstances, and have been suggested to be secondary to an immediate increase in pancreatic insulin release caused by GH.14
The anti-insulin activities of GH in animals were discovered many decades ago,15 when GH was found to inhibit the action of insulin, with associated rises in serum glucose levels. This activity was also shown in humans in the 1960s,16 and 19% to 56% of individuals with acromegaly develop type 2 diabetes that results from chronically elevated circulating insulin levels and subsequent insulin resistance. This increase in insulin results in an increased rate of triglyceride production, along with an altered lipoprotein profile.11
The anti-insulin effect of GH has been found to occur after relatively long periods of GH treatment, that is, after chronic exposure, both in cultured cells and in vivo, or in acromegalic patients who overproduce GH. This diabetogenic effect, which is thought to represent a major physiologic effect of GH, includes hyperglycemia secondary to an increase in hepatic glucose output following enhanced gluconeogenesis and glycolysis, hyperinsulinemia, and decreased glucose transport, as well as increased lipolysis. This last effect results in an increase in serum levels of nonesterified fatty acids, which further enhances the insulin-resistant state.10,17,18
The diabetogenic effect of GH is exerted directly by GH-induced intracellular signaling through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway in humans, as well as in rodents and cultured cell lines. Indeed, GH was recently shown to upregulate the p85α regulatory subunit of phosphoinositide (PI)-3 kinase expression and, thus, the activity of PI-3 kinase in white adipose tissue (WAT),19 skeletal muscle, and liver.20 This upregulation of p85α results in relative inhibition of the insulin signaling pathway, and ultimately insulin resistance accompanied by low levels of adiponectin, an insulin-sensitizing adipokine.19,20 However, other studies have questioned the role of PI-3 kinase in GH-induced insulin resistance in human muscle tissue.21
Mouse models of GH action have added significantly to the understanding of the physiologic effects of GH. These models include giant GH transgenic mice, dwarf GH antagonist (GHA) transgenic mice, and GHR gene deleted or knockout mice (GHR−/−) (Fig. 23-2). GH transgenic mice are giant, lean, and insulin resistant, and die prematurely as the result of kidney, liver, and heart problems. GHA transgenic mice are dwarf, have low levels of IGF-1, are somewhat insulin sensitive, and possess normal life spans. GHR−/− mice are dwarf and obese, express extremely low levels of IGF-1, are extremely insulin sensitive, and have extended longevity.22–24 The fact that GHR−/− mice are obese and yet insulin sensitive challenges the dogmatic notion that obesity is directly related to insulin resistance. However, contrary to what has been found in these mouse models, lipolysis induction by GH and its effect on body composition can indirectly exert a beneficial effect on insulin sensitivity, as is seen in GH-deficient patients treated with rGH.25 The effect of obesity on insulin resistance therefore may reside in the fat depot affected by GH.
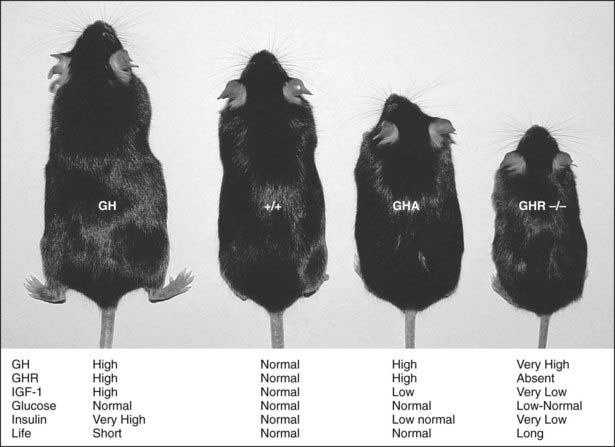
FIGURE 23-2. GH transgenic, wild-type (+/+), GH antagonist (GHA) transgenic, and GHR/BP gene-disrupted (GHR−/−) mice. A wild-type mouse is shown second from the left (+/+). The GHR/BP (−/−) mouse is approximately half the size of the normal, wild-type (+/+) mouse and is slightly smaller than the GHA transgenic mice. General endocrine values, including life span (life), are noted.
One of the major physiologic effects of GH is its influence on body composition and adipose tissue distribution. As stated above, GH transgenic mice are giant and possess a lean phenotype. In contrast, GHR−/− mice are dwarf and obese.26,27 It is surprising that a nonuniform distribution of adipose tissue in these mouse models was discovered. In GHR−/− mice, the subcutaneous and retroperitoneal depots are increased relative to control mice.26,28 Also, GHR−/− mice exhibit major decreases in the numbers of intraperitoneal adipocytes, whereas subcutaneous adipocyte number is increased relative to controls.26,28 This differential effect of GH on adipose depots is an exciting new finding in the GH field that may help to resolve the mechanisms of GH-induced insulin resistance.
Many of the functional effects of GH may result from the autocrine, paracrine, and endocrine actions of IGF-1, production of which is directly stimulated by GH, as well as other modulators. (IGF-1 action will be reviewed in another chapter of this volume.) Nevertheless, determining which of the GH-associated physiologic effects are the direct outcomes of GH action and which are caused indirectly by IGF-1 has been and continues to be an active area of research.
The non–growth-related roles of GH have widened the clinical utilization of recombinant (r)hGH. Initially, rhGH was indicated for GH-deficient children. In addition, rhGH is now indicated for growth promotion during childhood and adolescence in several conditions associated with growth impairment in the absence of GH deficiency. These indications include Turner’s and Prader-Willi syndromes, chronic renal insufficiency, SHOX gene defects, and children born small for gestational age (SGA) without catch-up growth. Two further indications—idiopathic short stature and Noonan’s syndrome—have been approved in the United States, but not in Europe, for specific brands of rhGH. In addition, rhGH treatment has been approved for GH-deficient adults, after its beneficial effects on body composition, metabolic parameters, and quality of life in these individuals were documented.29
Involvement of GH in cancer was, and still is, a controversial issue. Data from the Pfizer International Metabolic Database (KIMS) database provide no evidence that administration of rhGH to humans causes or promotes cancer.30 In addition, in a long-term study in mice and rats, administration of rGH had no effect on the incidence of cancer.31 However, Swanson and colleagues have shown that GH signaling is important in mouse prostate32 and mammary33 carcinogenesis. These investigators crossed the GHR−/− mouse with the C3(1)/TAg mouse, in which males develop prostatic intraepithelial neoplasia (PIN) and females develop mammary carcinomas driven by the large T antigen (TAg). (In both sexes, carcinogenesis is known to progress to invasive prostate carcinoma in a manner similar to the process observed in humans.) Progeny of this cross were genotyped, and TAg/GHR+/+ and TAg/GHR−/− mice were compared. In both prostate and mammary cancer models, carcinogenesis was significantly slowed in animals harboring TAg but lacking GHR (TAg/GHR−/−) compared with TAg mice expressing wild-type GHR (TAg/GHR+/+).
Swanson’s group has also shown that the spontaneous dwarf rat (SDR), which lacks GH as the result of a point mutation in the GH gene, is resistant to chemically induced mammary carcinogenesis or TAg-driven prostate cancer.34 This model is significant in that the SDR differs from the Sprague-Dawley rat only by this single point mutation. Exposure of the Sprague-Dawley rat to N-methyl-N-nitrosourea (MNU) is one of the most commonly used and thoroughly characterized models for human breast cancer, particularly for the ability of hormones to regulate tumor growth, and is considered to be an excellent model of human breast cancer. Finally, this group has reported that the SDR can be made vulnerable to MNU-induced mammary carcinogenesis by treatment with GH, and that once mammary tumors were established, cessation of GH treatment induced rapid and dramatic regression of mammary tumors.35
Through analysis of converging results from epidemiologic research and in vivo carcinogenesis models, Pollak and coworkers have established an association between the GH/IGF-1 axis and cancer, showing that high levels of circulating IGF-1 are associated with a modest increase in the risk for several common cancers, such as colorectal, prostate, and breast cancer.36 Based on these findings, experimental pharmacologic strategies that reduce IGF-1 receptor (IGF-1R) signaling are currently under development. In addition, when mice that express a GH antagonist were exposed to chemically induced breast cancer, significant suppression of the development of breast cancer was noted.37 Finally, a GH antagonist inhibited the growth of human meningiomas and colorectal and breast carcinomas in xenograft experiments in mice, suggesting this class of drugs as potential therapeutic agents through blockade of GHR-mediated signal transduction pathways.38
Recently, Lobie and colleagues have shown that hGH is synthesized at a number of extrapituitary sites, with autocrine hGH expression found in certain human carcinoma cell lines. This autocrine GH was found to stimulate survival, proliferation, migration, and invasion of human microvascular endothelial cells. Furthermore, recent studies have demonstrated that autocrine hGH is a wild-type orthotopically expressed oncogene for immortalized human mammary epithelial cells. Thus, autocrine and paracrine hGH may play a key role in angiogenic and lymphangiogenic processes in tumor neovascularization.39 It is important to note that the GH antagonist (described above) inhibits some of these processes.40 Thus, the association of GH with the initiation and progression of a variety of cancers is controversial, requiring further study before any conclusions can be drawn. Nonetheless, the GH antagonist (Pegvisomant) and any interventions that downregulate the GH-IGF axis may be possible treatment options for several types of cancer.
To explain these various GH actions, several hypotheses have been presented, including (1) the existence of multiple GHRs; (2) the presence of multiple “active centers” in the GH molecule; and (3) the presence of small, active GH fragments (≈90 fragments have been studied) with a variety of activities.41 Data related to these hypotheses are presented in the following section.
STRUCTURE OF GH
The crystal structure of the GH molecule, in particular, porcine (p)GH, was solved in 1987 (Fig. 23-3).42 GH was found to be an elongated molecule with approximate dimensions of 55 × 35 × 35 Å. The molecule contains four α helices, which are tightly packed as antiparallel bundles aligned in an up-up-down-down orientation, and contain 54% of the 191 amino acids of GH. The molecule also contains a “large loop” between residues 33 and 75, a “smaller loop” between residues 129 and 154, and a “small loop” located at the carboxyterminus.42 In 1992, the crystal structure of hGH, along with the GH binding protein (GHBP), was solved.43 Again, four GH α helices were detected: α helix I (residues 9 through 34), α helix II (residues 72 through 92), α helix III (residues 106 through 128), and α helix IV (residues 155 through 184). Two small mini-helices, residues 38 through 47 and 64 through 79, were also found in the large loop between α helices I and II.43
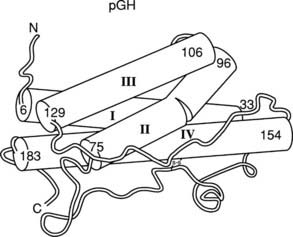
FIGURE 23-3. Crystal representation of porcine (pGH) at the 2.8 Å resolution level. Four α helices are depicted (cylindrical rods). The nonhelical region is shown as a thin tube. Also, one of the two disulfide bonds is shown; the other is hidden behind helix IV. The amino (A) and carboxyl (C) termini are located in the upper left and lower left corners, respectively.
(Modified from Abdel-Meguid SS, et al: Three-dimensional structure of a genetically engineered variant of porcine growth hormone, Proc Natl Acad Sci U S A 84[18]:6434–6437, 1987.)
The third α helix possesses amphiphilic characteristics, that is, the hydrophobic residues are geographically separated from the hydrophilic ones. Nevertheless, the third α helix of bovine (b)GH is imperfect in that Glu 117 (a hydophilic residue) is found in the hydrophobic one half of the α helix, while Ala 122 and Gly 119 (hydophobic residues) are positioned in the hydrophilic portion of the α helix. Amphiphilic secondary structures have been proposed to be important functional domains for many peptide hormones, as well as for transcriptional activators.
Bovine GH has four Cys residues located at positions 53, 164, 181, and 189, which are conserved among all GH, PRL, and placental lactogen molecules.6 The four Cys residues form two disulfide bridges in bGH (three in PRL) that are located between Cys 53 and Cys 164, which results in a large loop, and between Cys 181 and Cys 189, which forms a small C-terminal loop. Conservation of the Cys residues among members of the GH family may indicate that these residues are important for the structural integrity and biological activity of the molecules. Thus, disulfide bonds, as well as the third α helix, constitute potentially important elements for the activity of this molecule.
STRUCTURE/FUNCTION STUDIES OF GH
Multiple studies have been developed to assess the importance of the disulfide bonds in the activity of GH, including bond splitting and site-directed mutagenesis techniques targeting the Cys residues involved in disulfide bond formation. Results derived from these experiments were contrasting but suggested that the biological significance of the disulfide bond integrity may be species specific, and that the integrity of the large loop, but not that of the small loop, is essential for the growth-enhancing activity of GH.44 However, the effect of GH on lipid metabolism was unchanged.45
Information about functional domains of GH obtained through fragment experiments was limited because the overall conformation of the protein is not maintained. In the early 1990s, a novel approach toward understanding the structure of GH was employed using recombinant DNA techniques termed “homologue scanning.” Cloned DNA sequences encoding hPRL, which possess minimal GHR binding affinity, were substituted for corresponding regions of hGH, and the PRL/GH chimeric molecules were assayed for their ability to bind PRLR or GHR. This approach was very effective in defining the receptor binding domains of hGH.46 It was found that the GHR binding domains in hGH are located mainly in the NH2-terminal portion of α helix I, a loop region between amino acid residues 54 and 74, and the COOH-terminal portion of α helix IV.46 However, these experiments could not identify the specific residues involved in the ligand/receptor interaction.
Following the GH homologue scanning studies, a more refined approach was applied to the structure/binding relationships of GH and GHR. In this approach, Ala codons were systematically substituted for many codons in the GH gene, including those encoding residues found in α helix I, the large loop, and α helix IV. This “alanine scanning” approach was used to define specific amino acids residues important for GHR binding.47 It was reported that amino acid residues 10, 54, 56, 58, 64, and 68, which were in the loop region, and 171, 172, 175, 178, 182, and 185, in the C terminus, are involved in GHR binding.46,47 The scanning mutagenesis studies largely ignored the third α helix of GH because of the fact that amino acid substitutions in this region resulted in little change in receptor binding affinity.
THE THIRD α HELIX OF GH
The search for a growth-related domain in GH was pioneered by Sonenberg’s group in the late 60s and early 70s. Their main finding was that a short sequence, generated by tryptic digestion of bGH and containing residues 96 through 133, retained low but significant bone growth–stimulating activity, whereas segments 1 through 95 and 134 through 191 had much less activity. It is interesting to note that the tryptic peptide, 96-133, contains the third α helix of GH. Subsequently, it was reported that an hGH fragment (1-134) was fully active in an assay using the IM-9 strain of human lymphocyte assay.48
Recombinant hormones possessing different portions of GH, PRL, or PL also have been generated and analyzed. hGH 1-134 was linked to hPL 141-191 and hPL 1-134 was linked to hGH 141-191 through a Cys53-Cys165 disulfide bond.49 These recombinant hormones then were tested for both their immunoreactivities and their receptor binding properties; recombinant hGH (1-134)-hPL (141-191) retained hGH immunoreactivity and full GHR binding ability but had little hPL activity; on the other hand, recombinant hPL (1-134)-hGH (141-191) possessed a large quantity of hPL immunoreactivity and PRLR binding characteristics, with negligible hGH activity. These observations showed that the immunoreactivity and biological activity of hormones were determined primarily by the NH2-terminal fragment (residues 1 through 134), while the carboxyl-terminus appears to have little effect in determining the specificity of biological activity. Together these results suggested that GH activity could be ascribed to different regions of the GH molecule, and that the 96-133 segment might be an “active core” required for growth promotion.49
These two lines of evidence laid the foundation for the structure/function studies of the third α helix of GH. By combining site-specific mutagenesis of the GH gene with the ability of resulting bGH analogues to enhance the growth of transgenic mice, we have reported a growth-promoting region of GH localized in the third α helix (50-55), which is not a perfectly amphiphilic helix because of the presence of Glu 117, Gly 119, and Ala 122, as stated previously. To convert the imperfect third amphiphilic α helix of GH to a “perfectly amphiphilic” α helix, we substituted Glu 117, Gly 119, and Ala 122 with Leu, Arg, and Asp, respectively.50 The resulting GH analogue was bound to GHRs with the same affinity as native GH. However, when the Glu117Leu, Gly119Arg, Ala122Asp GH analogue (termed M8) was assayed for its ability to enhance growth in transgenic mice, this GH analogue did not enhance growth but suppressed it, resulting in mice with a dwarf phenotype. This was the first report of a GH analogue that antagonized the action of endogenous GH and, thus, the first report of a GHR antagonist.50
In a subsequent study, we extended this observation by performing individual amino acid substitutions. Substitution of Leu 117 for Glu resulted in a GH analogue that behaved identically to native GH,51 so we concluded that residue 117 of bGH is not likely to be involved in growth-promoting activity. In contrast, the bGH analogue Gly119Arg was found to bind to GHRs with the same affinity as native GH, but transgenic mice that expressed this analogue were about one-half the size of their non-transgenic littermates.51 This was the second report of a GHR antagonist. We further confirmed this observation by generating hGH-Gly120Arg dwarf transgenic mice.54 Also, several other amino acids were substituted for bGH Gly119 and were found to act as GHR antagonists.55 Finally, substitution of Asp for Ala at residue 122 results in a bGH analogue that binds to GHRs but does not enhance (or suppress) growth in transgenic mice and may be acting as a partial agonist. Together, these studies were the first to document the discovery of GHR antagonists.51–53
It is important to note that GH analogues with amino acid substitutions that resulted in changes in growth-promoting activity are located within a region of nine amino acids, that is, between Asp 115 and Leu 123,55 which form two turns of third helix. In viewing the side chains of these potentially important amino acids, it appears that Gly 119 and Ala 122, two amino acids with relatively small side chains, form a “hinge-like” or “cleft” structure that, as stated previously, has been shown to exist in the crystal structure of hGH,43 near the center of this α helix, primarily as the result of Gly 119 (Fig. 23-4). We postulated that this cleft is important for the growth-promoting activity of the GH molecule, and that Gly may be the only residue that is tolerable at this position.51 Extension of this model would yield the prediction that any other amino acid substitution at this position would decrease the flexibility of the molecule and/or “fill” the cleft, which ultimately would result in decreased biological activity. Finally, Asp 115 and Leu 123, amino acids with negatively charged (Asp) and long (Leu) side chains, respectively, flank the cleft and may be involved in the interaction with GHR.
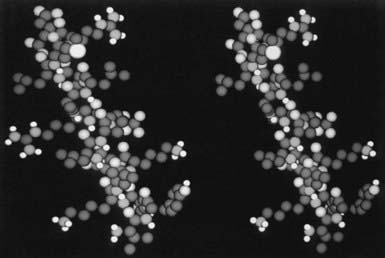
FIGURE 23-4. A space-filling model of the third α helix of GH (right) and a GH antagonist (left). The amino terminal of the helices is located on the top of the figure, and the COOH end at the bottom. Note the cleft that is located in the middle of the wild-type helix (right) and the occupancy of this cleft with the side chain of Arg (left).
(From Chen WY, et al: Glycine 119 of bovine growth hormone is critical for growth-promoting activity, Mol Endocrinol 5[12]:1845–1852, 1991.)
To further substantiate the importance of the cleft in the third α helix, we designed a bGH analogue with a deletion at Gly 119 (Δ119). Transgenic mice that expressed this analogue demonstrated a phenotype similar to that of their littermates.55 These data provide supportive evidence for the importance of the cleft structure in the third α helix. It is interesting to point out that all bGH analogues tested in this study were able to bind to the GHR with an affinity similar to bGH, including Δ119 (deletion mutation, inactive analog), SAP (scrambled helix, weak antagonist), and Gly119Arg (potent antagonist).
To accommodate all of the data related to amino acid substitutions in GH, including those derived from the alanine scanning studies47 and those directed at the third α helix of GH,50 we proposed the second target hypothesis for GH action.50 In this model, residues in α helices I and IV and the large loop region interact with the GHR, as reported by Cunningham.47 Additionally, we postulated that the cleft region in the third α helix interacts with an unidentified target, and the tripartite complex is the functional unit responsible for the induction of GH action.49
GH ANTAGONISTS
Gly 119 is conserved among all members of the GH family, including PRL and PL.6 Gly is unique among amino acids in that it possesses a single hydrogen atom as a side chain. The absolute conservation of this amino acid within a strong α-helical forming region of helix III of GH implies a crucial role for this residue.
As stated above, when bGH Gly 119 or hGH Gly 120 was replaced with a variety of amino acids and the mutated genes were expressed in transgenic mice, dwarf animals resulted.50,51,54 We also tested for the ability of the GH-substituted molecules to inhibit GH-dependent conversion of mouse preadipocytes to adipocytes. The bGH Gly119Arg or hGH Gly120Arg analogues were found to inhibit this reaction by 50% at equal molar concentrations of GH and analogues, thereby defining them as GHR antagonists.56,57 A confirmatory study on the generation of a GH antagonist by substitution of arginine for glycine at position 120 in hGH was subsequently reported.58
Later, Chihara and coworkers reported that another hGH gene mutation resulted in a “natural” GH antagonist.59 They also reported another hGH gene mutation that encoded an inactive GH60 in patients with reduced growth and short stature. In the first case, the codon for Arg 77 was found to be mutated so as to encode Cys, and the resulting molecule inhibited GH-stimulated JAK2 phosphorylation in vitro.59 Unexpectedly, the expression of this GH analogue, hGH Arg77Cys, in transgenic mice resulted in giant animals (Stevens and Kopchick, unpublished results). In the second case, the mutation resulted in the substitution of Gly for Asp acid at codon 112, found to be within Site 2 of the GH molecule60 (see later). No additional data have been reported on these GH gene mutations, which may encode a GHR antagonist and/or inactive GH.
In vitro and in vivo studies of hGH antagonists (GHAs) have demonstrated that they possess great potential to counteract the pathologic conditions of excess hGH in clinical settings, which include acromegaly, diabetic nephropathy, diabetic retinopathy, and, as stated before, certain cancers. Additionally, when GH (giant) and GHA (dwarf) mice are crossed, the resulting offspring are intermediate in size, suggesting that GHAs may overcome the growth-enhancing properties of GH.61
The GHA (hGH-Gly120Lys), like wild-type GH, has a short half-life,62 which limits its utility in the clinical setting. To increase the serum half-life of the molecule, the hGH antagonist has been modified by the addition of polyethylene-glycol (PEG); hGH-Gly120Lys with four to six PEGs has a half-life of approximately 18 hours after single intravenous (iv), intraperitoneal (ip), or subcutaneous (sc) injection.62 When mice received a daily sc single injection of various doses (0.25 to 4 mg/kg) of Gly120Lys-PEG or vehicle for 5 days, a significant, dose-dependent suppression of IGF-1 became obvious, starting at day 3. The maximum suppression (up to 70%) of IGF-1 production was achieved by 1 mg/kg dosing at day 6 after the first injection. Hepatic GHRs were significantly increased on day 8, also in a dose-dependent manner (Chen et al., unpublished data). These results suggest that exogenous administration of Gly120Lys-PEG can dramatically decrease serum IGF-1 levels.
These mouse data led to the development of the first hGH antagonist for the treatment of individuals with acromegaly. This hGH antagonist included eight amino acid substitutions at Site 1 of GH and the original G120K substitution at Site 2, along with four to five PEG additions. This molecule was termed B2036-PEG and was also called Pegvisomant. Pegylation of the molecule reduces clearance and therefore increases the serum half life and reduces immunogenicity, as well as its interaction with the GHBP.63 Thus, relatively high doses of Pegvisomant are required to lower serum IGF-1. Also, Pegvisomant binds to a pre-formed receptor dimer64 with an affinity similar to wild-type G120K and induces internalization but not subsequent GH-dependent intracellular signaling63,65 (Fig. 23-5). Thus, the importance of the eight amino acid substitutions in Pegvisomant is not one of increased GHR binding characteristics. However, because two of the amino acid substitutions at GH Site 1 involved Lys residues, and because Lys are potential pegylation sites, the importance of the changes may be that GH Site 1 cannot be pegylated. Thus, the molecule would be able to interact with the GHR.63
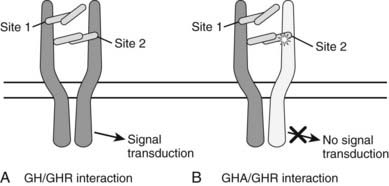
FIGURE 23-5. A, The one-GH/two-GHR model of GH action. The GH molecule and its four α helices are represented by the horizontal cylinders. The pre-formed GHR is shown as crossing the cellular membrane (dark horizontal lines). The interaction of GH with the pre-formed GHR at Site 1 and Site 2 is indicated. Not shown are the several N-linked glycosylation sites on the GHR. B, The interaction of a GH antagonist (A) with the pre-formed GHR dimer. The GHA and its four α helices are represented by horizontal cylinders. The Gly-to-Lys change in the third α helix is depicted by a star. The interaction of GHA with the pre-formed GHR at Site 1 and Site 2 is indicated. An improper or nonfunctional GHA/GHR dimer is indicated by the light shade of gray in the second GHR molecule found in the pre-formed dimer. Not shown are the several N-linked glycosylation sites on the GHR.
Debate has occurred over the mechanism of action of the GH antagonist. Fuh et al.58 suggested that the antagonist prevented the formation of the receptor dimer. However, because the GHR is pre-dimerized,63–65 the GHAs do not prevent GHR dimerization but prevent proper or functional GHR dimerization and subsequent signal transduction that ultimately results in decreased IGF-1 levels.66
Somavert (Pegvisomant for injection) has been approved for use in acromegalic individuals in the United States, Europe, and Japan. Data describing the results of Pegvisomant in acromegalic individuals, as well as their quality of life after treatment, have been put forth67,68 and as discussed in Chapter 14.
CO-CRYSTALLIZATION OF GH WITH THE GHR
As was stated previously, when the crystal structure of hGH complexed with GHBP was solved,43 two identical GHBP molecules were found to interact with one GH molecule. This observation of two GHRs interacting with one GH molecule (Fig. 23-6) is one of the most fundamental findings in the GH molecular endocrinology field. Thus, the co-crystallization of one GH molecule with two GHBPs supported the GH second target hypothesis of GH action,50 that is, the “second target” was another GHR. It should be pointed out that reagents used in these co-crystallization studies include non-glycosylated, bacterially synthesized GHBP, and not a membrane-associated and glycosylated full-length GHR. Also, GH has not been found in a GHBP dimer in vivo.
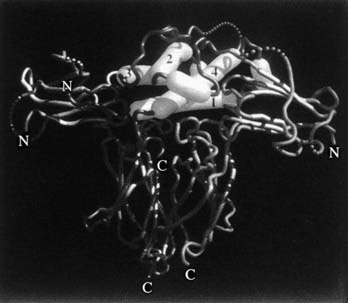
FIGURE 23-6. Representation of the GH/GHR co-crystal structure. The GH α helices are indicated as cylinders and are labeled 1, 2, 3, and 4. α helices 1 and 4 (Site 1) are shown interacting with one GHBP; α helix 3 (Site 2) is shown interacting with a second GHBP.
(From de Vos AM, Ultsch M, Kossiakoff AA: Human growth hormone and extracellular domain of its receptor: crystal structure of the complex, Science 255[5042]:306–312, 1992.)
Growth Hormone Receptor
GHR AND GHBP
Growth hormone receptors (GHRs) have been found on the cell surfaces of many tissues throughout the body, including liver, muscle, adipose, and kidney, and in early embryonic and fetal tissue. Although most GHRs reside on the cell surface and in the endoplasmic reticulum, pronounced nuclear localization is noted in many cells.69 Evidence for the importance of the GHR in growth has come from studies on individuals expressing different mutations located throughout the GHR gene that result in the dwarf phenotype and a GH-insensitive state, also termed Laron syndrome. Later, another proof of the importance of the GHR in growth was shown with disruption of the GHR and GHBP genes in mice.70 These mice are approximately half the size of wild-type mice and are obese; they express very low levels of IGF-1 and insulin and high levels of GH, and they have an extended life span.24,71 Whether data on extended life span from GHR-/- mice extends to humans is not known. More research on this controversial subject will ensue.
The GHR is a member of the class 1 hematopoietic cytokine family. The human GHR gene encompasses 10 exons and approximately 90 kb, and encodes an extracellular domain, a small transmembrane domain, and an intracellular domain. The protein-coding region of the GHR gene is encoded by exons 2 through 10. Exon 2 of the GHR gene encodes the secretory signal peptide and the first six amino acids of the mature form of the protein; exons 3 through 7 encode the extracellular domain, exon 8 the transmembrane domain, and exons 9 and 10 the intracellular domain.
The extracellular portion of the GHR consists of two fibronectin type III domains, each containing seven β-strands, arranged to form a sandwich of two antiparallel β-sheets.43 Stabilizing the GHR structure is a salt bridge between Arg 39 and Asp 132, and hydrogen bonds between Arg 43 and Glu 169.43 Also, the GHR contains seven cysteine residues in its extracellular domain43; the six in the GH binding domain form three disulfide bonds in the active signaling conformation, and help the receptor to maintain its correct three-dimensional structure.72 Van den Eijnden and coworkers have suggested, after studying the effects of replacing the Cys with Ser and Ala residues, that the middle disulfide bond, Cys83-Cys94, is important for ligand binding, whereas removal of disulfide bond Cys108-Cys122 has little effect on GH-induced intracellular signaling.72 The GHR has a half-life of approximately 1 hour and is degraded continuously even in the absence of GH through two known mechanisms: endocytosis and ectodomain cleavage. The reader is referred to excellent reviews on the GHR.69,73,74
In addition to the membrane-bound GHR, a soluble form exists that is composed of a portion of the extracellular domain, GHBP. In mice and rats, it is encoded by an additional exon, Exon 8A, and is produced by alternative splicing of the GHR precursor mRNA. In other vertebrates, it is believed that it is generated by proteolytic cleavage of the extracellular domain of the GHR. A metalloprotease tumor necrosis factor (TNF)-α converting enzyme (TACE/ADAM-17) acts on surface GHR to generate the GHBP.75 The function of the GHBP is not fully understood, but it may modulate GH activity by enhancing its half-life or reducing its availability to bind the GHR. The reader is referred to a review paper on GHBPs for further information.76
GH/GHR INTERACTION
Examination of the 2.8 Å crystal structure of the complex between GH and the extracellular domain of the GHR produced by Escherichia coli (hGHBP) demonstrated that the complex consisted of one molecule of GH and two molecules of the GHR (Fig. 23-6).43 Furthermore, the crystal structure reveals how a nonsymmetrical molecule, that is, GH, binds to two copies of the GHR, with one of the GHRs binding with high affinity to the GH molecule at Site 1, and with a weaker binding between Site 2 of GH and a second GHR.
Amino acid residues in the hGHR (actually hGHBP) involved in contact with hGH have been determined from co-crystallization analyses of the GH/GHR complex.43 The major binding determinants in the GH molecule (Site 1), located in the two mini-helices between α helices 1 and 2 (amino acids 60 through 63) and between the center and carboxyterminus of helix IV (amino acids 168 through 174), match to GHR amino acids 40 through 45 and 101 through 106, respectively, and with GHR Trp 169 interacting with hGH residues 171 through 179 in α helix IV. Site 2 GH residues are important for contact and dimerization and only Phe 1, Ile 4, and Asp 116, whereas the significant binding determinants in the GHR are similar as for GHR Site 1, especially Trp 104 and Trp 109.77 Of particular interest is the close encounter with hGH Gly 120 and GHR Trp 104 in this Site 2 interaction.
The model for GHR activation postulates that GH induces GHR dimerization and, consequently, activation and signaling in which most amino acid residues in the two binding interfaces act in an additive fashion.78 In addition, the GHR contains a GH-induced dimerization domain in which Cys 241 undergoes GH-induced intermolecular disulfide bonding, thus bridging together two GHRs. Eight hGHR amino acid residues are involved in the salt bridge and the hydrogen bond interactions across the extracellular dimerization domain.43 Of these eight residues, five are important for GH/GHR-mediated signal transduction, namely, Ser 145, His 150, Asp 152, Try 200, and Ser 201, but not Leu 146 or Thr 147. This study, as well as others using monoclonal antibodies to induce a GH response,79 suggests that a GH-induced conformational change in the GHR is required for a full biological response. Additionally, subtle but significant differences between the 1hGH/2GHR43 and 1hGH/1GHR80 co-crystal structures suggest that a conformational change does occur in the one ligand/two receptor complex.
Although most if not all of these GH/GHBP interactive studies use a non-glycosylated, bacterially expressed GHBP, and not the membrane bound GHR, the interaction of one GH with two GHBPs has been extrapolated to the in vivo interaction of GH with the GHR. This finding has led to the theory of a sequential binding mechanism in which hGH binds to two GHRs.77 In this model, hGH must first bind to one GHR using a high-affinity receptor binding site, which subsequently allows binding of the second receptor. The binding Site 1 of hGH is located at residues identified by Ala scanning mutagenesis studies,47 that is, α helix I, the loop between amino acids 54 through 74 and α helix four. Binding Site 2 is located at the N-terminus (Ile 4), and the third α helix, namely, Gly 120. The model predicts that a Typ 104 residue of the GHR “fits” into the “cleft” of the third α helix of GH. Chen et al.56,57 have proposed that the reason that hGH-Gly120Arg acts as a GH antagonist is because substitution of Arg for Gly at position 120 blocked or inhibited the “second” GHR from properly interacting with binding Site 2 on the GH molecule. This would inhibit proper or functional GH-induced GHR dimerization. These data nicely supported the “cleft” theory pertaining to the interaction of hGH Gly 120 with a second target.56,57 The importance of GH-induced GHR dimerization in humans has been supported by the finding of an adenine-to-guanine mutation in the hGH gene that results in the conversion of Asp112 to Gly. In the heterozygous state, this mutation is believed to be the cause of dwarfism in a female child, with the encoded hGH analogue binding to hGHR in vitro, but not inducing GHR dimerization and JAK2 and STAT5 activation.60
Later studies have dramatically changed the GH-induced dimerization theory. The work of Gent et al.,64 who used coimmunoprecipitation and epitope-tagged truncated GHRs, conclusively showed that ligand-independent GHR dimerization occurs in the endoplasmic reticulum and on cell membranes independent of GH binding.64 In addition, studies with GHAs (bGH Gly119Arg, hGH Gly120Lys; B2036 and Pegvisomant) have demonstrated that these antagonists exist in a complex with two GHRs and are internalized properly.63,65 Thus, results showed that GHAs prevent neither GHR dimerization nor internalization, but they interfere with proper GHR dimerization. This has led to the proposed model of GH binding to a constitutively homodimerized GHR, causing a structural change that results in activation of JAK2 and signal transduction.69
The mechanism by which GH binding converts the inactive pre-dimerized GHR to its active conformation remains uncertain. However, it has been shown that the composition and/or length of the transmembrane domain does not affect GH-induced GHR dimerization.81 Also, through the mutation of Site 2 in GHR, this site has been shown to be not essential for GHR to achieve its active conformation (identified by GHR disulfide linkages) and to trigger signal transduction. Additionally, the extracellular domain of GHR shows substantial flexibility to achieve active conformation in response to GH and will even accommodate GH-GH dimers.82 The interaction of GH with the GHR has been proposed to cause repositioning of the intracellular domain of GHR, in which rotation of individual GHR molecules ultimately triggers GH-induced intracellular signal transduction.83 This model, developed by Waters and colleagues over the past few years, has greatly influenced and stimulated research on the interaction of GH with GHRs and represents a seminal finding in the GH field.83
Finally, the mechanisms responsible for GHR turnover (proteolysis) have been shown to influence GH sensitivity. As was stated before, cleavage of the extracellular domain of the GHR is catalyzed by a transmembrane ectoenzyme termed TACE (tumor necrosis-α cleaving enzyme, also known as ADAM-17).75 GHR proteolysis renders cells less sensitive to subsequent GH-induced signaling by downregulating GHR abundance. Recent in vivo studies indicate that this mechanism of receptor downregulation may also mediate desensitization to GH in vivo84 and in pathophysiologic states in which it may be advantageous to limit the anabolic effects of GH (e.g., sepsis). It is interesting to note that the GHR remnant that results from metalloproteolysis is further cleaved within the transmembrane domain by an enzyme termed γ-secretase, leading to release of the intracellular domain into the cytosol and its accumulation in the nucleus. The consequences of this inducible nuclear localization of the GHR intracellular domain are as yet unknown, but may suggest novel GHR-dependent signaling pathways.
We have only touched on a few of the structural characteristics of the GHR. For a more exhaustive review of this subject, the reader is referred to an excellent review by Brooks, Waters, and coworkers.69
CLINICAL MANISFESTATIONS OF MUTATIONS IN THE GHR
GH-insensitive individuals with a dwarf phenotype were first described by Laron. The molecular defect in these Laron syndrome (LS) individuals has been found in the GHR. Since the initial series of GHR mutations was documented in LS individuals, many other sites of GHR mutation have been documented.85 Because these individuals are GH insensitive, treatment with IGF-1 is the only option.85,86
In 2004, Dos Santos et al.87 reported that a rather frequent polymorphism of the GHR gene was associated with increased responsiveness to GH. This GHR gene mutation resulted in removal of exon 3 and has been termed the d3 GHR allele.87 Children born SGA, children with idiopathic short stature, girls with Turner’s syndrome, and children with severe GH deficiency all in the presence of a d3 GHR polymorphism have a greater response to exogenous rhGH administration than do similar individuals who express the full-length GHR. These findings suggest that a subpopulation of individuals with the d3 GHR allele may have increased sensitivity to GH. Also, in a study of acromegalic individuals,88 the wild-type, full-length GHR was found to be homozygous in 50% of patients, while 50% had at least one d3 GHR allele. The GHR genotype (specifically, the deletion of exon 3) was hypothesized to modulate the relationship between GH and IGF-1 serum concentrations in these acromegalic individuals.88
GH-Induced Signal Transduction
The molecular mechanisms by which GH transmits its signals via its receptor have been largely elucidated by experiments in cultured cells or hypophysectomized rats. However, GH-induced in vivo tissue-specific signal transduction systems are still largely unknown, although data are beginning to emerge in this area, as is knowledge about modulators of GHR.21,89,90 Several GH-mediated intracellular signal transduction pathways are summarized below, some of which may overlap with signal transduction intermediates induced by insulin and other hormones, thus providing opportunities for “biological cross-talk” between these molecules. Several reviews on the subject have been presented.89,90 It is interesting to note that one of the hormones involved in this cross-talk is GH-induced IGF-1. Recently, examination of GH signaling in murine 3T3-F442A pre-adipocytes revealed that GH induces formation of a complex that includes the GHR, JAK2, and the IGF-1 receptor (IGF-1R).91 Even though both the GHR and JAK2 in the complex are tyrosine phosphorylated, formation of this complex does not depend on tyrosine phosphorylation of any of the partners. It is interesting to note that co-treatment of cells with the combination of GH and IGF-1 resulted in enhancement of downstream signals compared with GH alone. This suggests functional consequences of the complex formation. Further evidence for the idea that the GHR and the IGF-1R may function in parallel has come from studies in primary mouse osteoblasts in which the IGF-1R gene is flanked by loxP sites that enable excision of the gene when Cre recombinase is expressed. When IGF-1R was deleted from these cells, GH-induced STAT5 activation was substantially diminished, suggesting that the IGF-1R, even when not engaged by IGF-1, contributes to optimal GH-induced GHR signaling.92
JANUS KINASE ACTIVATION
In the early 1990s, GH treatment of responsive cells was found to induce association of a tyrosine kinase with the GHR. This kinase was later identified as a 121 kDA protein and was found to be a member of the Janus kinase (JAK) family of proteins, in particular JAK2. Activation of JAKs appears to be an initial step in one of the GH-induced signal transduction systems. Although three JAK molecules are involved in GH/GHR signal transduction, JAK2 exhibits the greatest degree of activation. GH-dependent JAK2 activation requires interaction between JAK2 and the membrane-proximal, proline-rich motif (termed Box 1) located in the intracellular region of GHR. Because the GHR itself has no intrinsic kinase activity, it is thought that co-localization of two JAK2 molecules by the dimerized GHR results in transphosphorylation of one JAK2 by the other, leading to JAK2 activation. Activated JAK2, in turn, is thought to phosphorylate GHR on multiple tyrosine residues, providing docking sites for cytosolic components of at least three distinct signaling pathways: the STAT, MAPK, and PI3K pathways.90,93,94 An overview of these pathways is presented in Fig. 23-7.
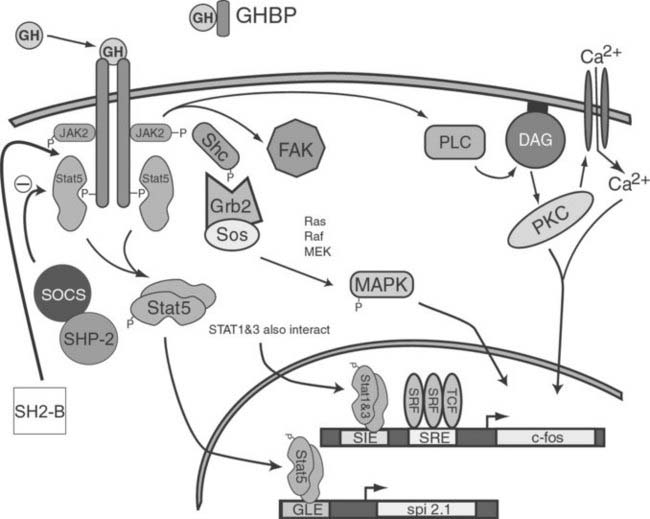
FIGURE 23-7. A model depicting intracellular signaling intermediates induced by binding of GH with the GHR.
(Modified from Kopchick JJ, Andry JM: Growth hormone [GH], GH receptor, and signal transduction, Mol Genet Metab 71[1-2]:293–314, 2000.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


