FIGURE 107-1. Schematic representation of the renin-angiotensin-aldosterone system in primary aldosteronism. Aldosterone secretion is increased independently of the renin-angiotensin system. Aldosterone increases sodium (Na+) reabsorption at the renal cortical collecting duct in exchange for potassium (K+) and hydrogen (H+) ions. Reabsorbed Na+ increases extracellular fluid volume, inhibiting renin production and secretion. For this reason, angiotensin peptide synthesis is suppressed. K+, normally a stimulator of aldosterone production, is reduced, leading to partial reduction of autonomous aldosterone secretion. Reduced activity of the renin-angiotensin system and K+ denoted by gray.
Following the original description, identical clinical and biochemical features were discovered in patients with bilateral disease without adrenal tumors. The spectrum of primary aldosteronism as currently recognized is listed in Table 107-1, together with estimates of the prevalence of its different subtypes. In addition to these disorders, the clinical diagnosis of primary aldosteronism should be distinguished from administration of exogenous mineralocorticoids (e.g., fludrocortisone) or drugs or substances previously thought to be mineralocorticoids but now known to act indirectly via inhibition of 11β-hydroxysteroid dehydrogenase (11β-HSD) (e.g., licorice, carbenoxolone) or secretion of excess mineralocorticoids other than aldosterone (e.g., deoxycorticosterone [DOC]).6
Table 107-1. Forms of Primary Aldosteronism and Their Prevalence Rates
| Disorder | Prevalence, % |
|---|---|
| Aldosterone-producing adenoma (APA) | 33 |
| Bilateral idiopathic adrenal hyperplasia (IHA) | 63 |
| Primary (unilateral) adrenal hyperplasia | <1 |
| Aldosterone-producing adrenocortical carcinoma | <1 |
| Ectopic (nonadrenal) aldosterone-producing adenomas | Rare |
| Familial hyperaldosteronism (FH) | |
| Type I (FH I)—Glucocorticoid-remediable hyperaldosteronism | 0.5 |
| Type II (FH II)—Adrenocorticotropic hormone (ACTH)-independent familial hyperaldosteronism | 3-4 |
Definition of Primary Aldosteronism
As stated in the 2008 Endocrine Society Clinical Practice Guideline,7 primary aldosteronism is defined as a group of disorders in which aldosterone production is inappropriately high, relatively autonomous, and independent of the renin-angiotensin system, and in which aldosterone secretion is not suppressed by sodium loading. Hypokalemia was formerly included as a part of the definition of primary aldosteronism. However, in recent studies in unselected hypertensive populations, most patients with primary aldosteronism have been normokalemic. Rarely, patients with primary aldosteronism are normotensive, and very rarely normal levels of plasma and urinary aldosterone have been reported.8,9
Prevalence of Primary Aldosteronism
Conn10 originally believed that approximately 20% of patients with essential hypertension might have the syndrome that he had originally reported. At the time, this was thought to be a gross overestimate of prevalence. Later, Conn adjusted his estimated prevalence to approximately 10% of hypertensive individuals—a prediction that has been substantiated approximately 40 years later. Until the early to mid 1980s, when routine measurement of the aldosterone:renin ratio was introduced, most authors suggested that the prevalence of primary aldosteronism was less than 1% in unselected hypertensive populations.11–14 For example, Lewin and collegues13 identified only 3 of 5485 patients with possible primary aldosteronism. A slightly higher prevalence (0.4%) was found in 3783 patients with moderately severe, nonmalignant hypertension in Glasgow.14 Other investigators, however, found a significantly higher prevalence when studying selected populations, especially when aldosterone:renin ratios were employed for screening all patients with hypertension and not simply those with hypokalemia (2% to 12%).15–18
Use of the plasma aldosterone concentration (PAC):plasma renin activity (PRA) ratio as a screening test followed by aldosterone suppression confirmatory testing to make the definitive diagnosis has led to much higher prevalence rates of primary aldosteronism in patients from five continents.17 With use of these methods, the prevalence of primary aldosteronism now is generally accepted as 5% to 13% of all patients with hypertension.17–26
Cause of Primary Aldosteronism
In addition to aldosterone-producing adenoma (APA) as described by Conn, six subtypes of primary aldosteronism have been described (see Table 107-1). With the exception of glucocorticoid-remediable aldosteronism (GRA) (also termed familial hyperaldosteronism type I [FH-I], dexamethasone-suppressible aldosteronism, or glucocorticoid-remediable hyperaldosteronism), the precise molecular origin of primary aldosteronism has not been determined.
GRA is an autosomal dominant disorder that is responsible for less than 1% of cases of primary aldosteronsim.27 GRA is characterized by the early onset of moderate to severe hypertension, premature cerebrovascular accidents, variable hypokalemia (most patients are normokalemic), aldosterone excess with suppression of PRA, and excess production of the hybrid steroids 18-hydroxycortisol (18-OH-F) and 18-oxocortisol (18-oxo-F).27 GRA is caused by a novel gene duplication (i.e., a chimeric gene) resulting from unequal crossing over between the promoter sequence of the CYP11β1 gene encoding 11β-hydroxylase and the coding sequence of the CYP11β2 gene encoding aldosterone synthase.27,28 This gene duplication allows ectopic expression of the aldosterone synthase enzyme in the adrenal zona fasciculata, where it is not normally expressed, explaining the adrenocorticotropic hormone (ACTH)-dependent aldosterone overproduction and the increased levels of cortisol hybrid steroids.27,28 The steroid secretion pattern found in GRA is compared with the normal pattern in Fig. 107-2. The fact that patients with GRA are frequently normokalemic despite clear aldosterone excess is consistent with findings of recent studies involving patients with other forms of primary aldosteronism, most of whom are also normokalemic.17,26 Because of similarities between GRA and angiotensin-unresponsive aldosterone-producing adenomas (APAs) (the primary regulation of aldosterone is provided by ACTH), investigators have tested whether the chimeric gene found in GRA is present in APA, but it is not.29 An abnormality of aldosterone synthase expression or activity has not been reported in APA or IHA.
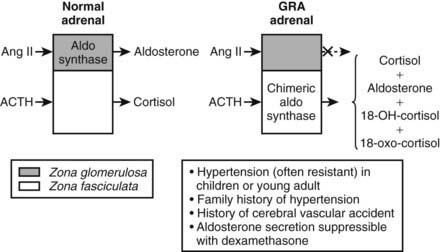
FIGURE 107-2. Schematic depiction of the pathophysiology and major clinical manifestations of glucocorticoid-remediable alsteronism (GRA). Dashed arrow indicates response absent.
A different form of familial primary aldosteronism, which also has an autosomal dominant inheritance pattern and may have a monogenic basis, is familial hyperaldosteronism type II (FH-II).30 In this disorder, aldosterone is not suppressible by glucocorticoids, and no hybrid gene mutation occurs. FH-II can present with APA or adrenocortical hyperplasia. In the extensive experience of the Brisbane, Australia group, FH-II is more common (3% to 4%) than FH-I, and the diagnosis is made at an older age than for FH-I. No difference in age, gender, biochemical parameters, or aldosterone or renin levels has been documented between FH-II and sporadic primary aldosteronism. The molecular basis for FH-II is unclear. However, linkage analysis in large families has recently shown an association with chromosome region 7p22.30,31 Possible candidate genes at this locus include PPKAR1β encoding the 1-β regulatory subunit of protein kinase A. A related gene encoding the 1-α regulatory subunit, PPKR1α is mutated in the Carney complex, another familial condition associated with adrenal cortisol-producing tumors.
Idiopathic hyperaldosteronism (IHA), the most common form of primary aldosteronism, is characterized by bilateral adrenal hyperplasia. The cause of IHA is unknown. Adrenals may be normal or may display diffuse, micronodular, or macronodular hyperplasia. Available evidence suggests that the aldosterone overproduction is angiotensin II dependent and may result at least in part from hypersensitivity of the zona glomerulosa to angiotensin II.32,33 The positive interrelationship of plasma aldosterone to angiotensin II suggests that IHA may be a form of secondary rather than primary hyperaldosteronism. Padfield and associates33 suggested that IHA is part of a continuum with low renin essential hypertension (LREH). However, the fact that aldosterone secretion is at least partially autonomous of the renin-angiotensin system in IHA, as demonstrated by failure of normal suppression during sodium loading (despite profound suppression of renin), argues against this possibility. Wisgerhoff and associates34,35 first documented the exaggerated angiotensin II–induced aldosterone response in both IHA and LREH. Witzgall and colleagues36 later confirmed increased aldosterone hypersensitivity to angiotensin II and in separate experiments determined that there was increased but ineffective dopaminergic inhibition of aldosterone secretion in both IHA and LREH. However, another group failed to confirm this observation.37
The role of angiotensin II in stimulating aldosterone secretion in IHA is supported by the ability of captopril and other angiotensin-converting enzyme (ACE) inhibitors to reduce plasma aldosterone in this condition. The source of the circulating angiotensin II, the synthesis of which is interrupted by converting enzyme inhibition, is unclear in the presence of low PRA. This observation has raised the possibility of an abnormality of the intra-adrenal (local tissue) renin-angiotensin system independent of the systemic circulation. Fallo and coworkers38 examined the effects of captopril on aldosterone responses to potassium infusion in IHA and APA. Before captopril, potassium stimulated an increase in aldosterone in both groups. After captopril, the response was significantly blunted in IHA but not in APA. These observations suggested that the intra-adrenal renin-angiotensin system may play a role in the aldosterone response to potassium in IHA. However, direct in vitro evidence for this hypothesis is lacking.
Klemm and colleagues39 reported an increase in renin gene expression in angiotensin-responsive APAs (which biochemically behave similarly to IHA in that aldosterone production is responsive to angiotensin) but not in angiotensin-unresponsive APAs compared with normal tissue. Renin gene expression was also increased in adrenal cortex surrounding some angiotensin-responsive APAs, suggesting that the defect in tissue renin gene expression may not be confined to the tumor.
Although several investigators have attempted to implicate pituitary peptides other than ACTH, especially pro-opiomelanocortin derivatives, no consistent evidence for this hypothesis has been forthcoming in the pathophysiology of IHA.40
Essentially nothing is known about the cause of primary adrenal hyperplasia (PAH) in which both adrenal glands or, more rarely, one adrenal gland shows micronodular or macronodular hyperplasia and produces a clinical and biochemical picture very similar to that of angiotensin-unresponsive APA.41,42
Recently, another variety of familial primary aldosteronism featuring “non–glucocorticoid-remediable aldosteronism” was described in a father and two daughters.43 The patients all had increased circulating levels of 18-oxocortisol and 18-hydroxycortisol, steroids reflecting oxidation by both 17-hydroxylase and aldosterone synthase and administration of dexamethasone failed to suppress aldosterone secretion. Bilateral adrenal hyperplasia was found at surgery and, in contrast to IHA, bilateral adrenalectomy corrected the hypertension.43 Genetic information has not been reported for these patients.
Until 2008, no animal models of IHA were available for study. Davies and colleagues44 have now established an animal model of nontumorigenic primary aldosteronism via deletion of TWIK-related acid-sensitive K (TASK)-1 and TASK-3 channels in mice. This deletion removes an important adrenal background K+ currrent, which results in marked depolarization of zona glomerulosa cell membrane potential. Double–TASK channel knockout mice exhibited overproduction of aldosterone, suppressed renin, lack of salt suppressibility of aldosterone secretion, and failure of angiotensin type-1 (AT1) receptor blockade to lower aldosterone to control levels. Thus, these mice exhibit a phenotype similar to that of IHA.44 The interesting possibility that human IHA is related to mutations in TASK channel genes awaits future investigation.
Pathobiology of Primary Aldosteronism
Three major pathologic disorders are associated with primary aldosteronism: adenoma, hyperplasia, and carcinoma.
Adrenocortical Adenoma
Adenoma, which is found in approximately 60% of patients with primary aldosteronism, was once considered to be the most common abnormality. However, with the current use of PA:PRA ratios for case detection, IHA is now the most common subtype (63%). This change in subtype prevalence is almost certainly related to the less florid clinical and biochemical manifestations of IHA compared with APA, together with the current ability of sensitive screening methods to detect patients with IHA.
Adrenal adenomas occur somewhat more frequently in the left than in the right adrenal.2,45 Adenomas usually measure less than 2 cm in diameter, and have a golden yellow color (Fig. 107-3). On light microscopy, four cell types have been identified: small and large hybrid cells with features of both zona glomerulosa and zona fasciculata cells, and others with zona glomerulosa or zona fasciculata characteristics.46 Electron microscopy has shown that most of the mitochondria possess tubular cristae similar to those found in the cells of the zona glomerulosa. If the patient has been exposed to spironolactone therapy, spironolactone bodies may be seen.46,47 It is interesting to note that zona glomerulosa hyperplasia often accompanies APA in the surrounding cortex.46 In unusual circumstances, multiple adenomas or an adenoma with associated macronodular hyperplasia or smaller satellite nodules can be found. These findings, which are similar to those of multiple endocrine neoplasia, have suggested that genetic mutations affecting regulation of adrenocortical cell growth and differentiation may serve as the basis for at least some cases of primary aldosteronism.48 It is interesting that a patient with bilateral adenomas with two types of adenoma cells associated with both primary aldosteronism and Cushing’s syndrome has also been reported.49
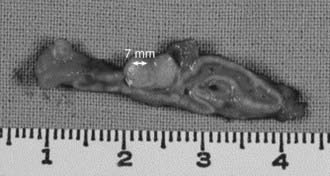
FIGURE 107-3. Typical aldosterone-producing adrenal adenoma removed from a patient with a 24-year history of resistant hypertension.
(From Ref. 111.)
Idiopathic Hyperaldosteronism
In IHA, the zona glomerulosa usually demonstrates diffuse or focal hyperplasia with normal ultrastructure but may be macroscopically normal.47,50 Associated nodules may be microscopic or may be as large as 2 cm in diameter, and their ultrastructure is typical of clear cells of zona fasciculata origin. In keeping with this observation, in vitro the nodules produce cortisol but not aldosterone. Immunohistochemistry for aldosterone synthase has demonstrated intense staining in the zona glomerulosa and outer zona fasciculata in IHA. The compact cells in APA also stain for aldosterone synthase.51
Adrenal pathologic findings may be important to the outcome resulting from unilateral adrenalectomy. Ito and colleagues52 studied 37 patients with primary aldosteronism: 23 had unilateral solitary adenomas (Group 1), 3 had unilateral multiple adenomas (Group 2), and 11 had adenomas with multiple macroscopic or microscopic nodules (Group 3). Postoperative changes in the renin-angiotensin-aldosterone system were similar, but marked differences in blood pressure responses were noted, as half of Group 3 remained hypertensive at 1 year. The authors suggested that adrenal nodules might result from long-standing hypertension. Glands with nodules almost invariably show arteriopathy of the capsular vessels, which may lead to focal ischemia and atrophy, with the better perfused cells becoming hyperplastic, leading to nodule formation.53
Adrenal Carcinoma
Adrenal carcinoma is a rare cause of primary aldosteronism. Histologically, carcinomas may be difficult to distinguish from adenomas, but almost invariably, carcinomas are larger (>3 cm in diameter) and include areas of necrosis and pleomorphic nuclei.46 Calcification, commonly found in carcinomas, may be detected by magnetic resonance imaging (MRI), computed tomography (CT) scanning, or ultrasonography.
Ectopic Aldosterone-Secreting Adenoma
Ectopic aldosterone-secreting adenoma is an extremely rare cause of primary aldosterone excess.53–57 Rarely, cases of primary aldosteronism have been reported in association with malignant ovarian tumors.53–56 After excision of the tumor, biochemical abnormalities and hypertension may resolve or improve. Recurrence of the ectopic tumor can produce a return of the syndrome.54
Clinical Presentation of Primary Aldosteronism
The clinical features of primary aldosteronism have been extensively described (Table 107-2).58–62 Most patients are asymptomatic. Some are discovered to be hypokalemic on routine investigation of hypertension, whereas others may have symptoms of hypokalemia such as muscle weakness, very occasional muscle paralysis, or more commonly, polyuria, polydipsia, nocturia (secondary to nephrogenic diabetes insipidus), paresthesias, and, rarely, tetany.63 Chinese patients have a high prevalence of hypokalemic periodic paralysis.64
Table 107-2. Clinical Manifestations of Primary Aldosteronism
Conn et al.65 found an intriguing sex difference in the prevalence of paresthesias and tetany in patients with primary aldosteronism; that is, females are more likely than males to develop paresthesias or to present with tetany. Tetany results from reduced ionized calcium accompanied by hypokalemic alkalosis. Plasma total calcium and magnesium levels are normal, however, and treatment consists of potassium repletion, not administration of calcium or magnesium.
Malignant hypertension originally was thought not to occur in primary aldosteronism.2 However, this was found to be erroneous, and many cases have now been reported.66,67 The diagnosis of primary aldosteronism can be missed in malignant hypertension because PRA may not be suppressed. On the other hand, normotensive aldosteronism has been documented.68,69 Normotensive primary aldosteronism is most likely to occur in families with familial hyperaldosteronism (FH-I or FH-II), as affected individuals may be detected at an early, preclinical stage of the disease process by means of genetic family screening programs.70
In primary aldosteronism, the normal circadian pattern of blood pressure appears to be preserved with nocturnal dipping,71 but the magnitude of the nighttime decrease is reduced.72 However, blood pressure variability may be reduced in primary aldosteronism compared with essential hypertension,73 possibly as a result of protection of baroreflex function, or as a consequence of the salt-loaded state with extracellular fluid volume expansion.
Originally, primary aldosteronism was thought to be a relatively benign disorder with low morbidity and mortality. Studies conducted during the past decade, however, have provided new insights into the role of aldosterone in tissue damage (inflammation, fibrosis, and remodeling) (Fig. 107-4).74 Many studies have shown that patients with primary aldosteronism are at higher risk than other patients with hypertension for target organ damage, especially involving the heart, kidneys, and blood vessels (Table 107-3).75–83 When matched for age, blood pressure, and duration of hypertension, patients with primary aldosteronism have greater left ventricular mass measurements compared with other patients with hypertension.77 In patients with APA, both left ventricular wall thickness and mass regress markedly within a year of unilateral adrenalectomy. Patients who present with APA or IHA have an increased number of cardiovascular events when compared with an age-, gender- and blood pressure–matched population with essential hypertension.75 In primary aldosteronism, a striking increase in the relative risks for stroke (4.2%), myocardial infarction (6.5%), and atrial fibrillation (12.1%) has been noted.75 Patients with primary aldosteronism have enhanced diastolic dysfunction when compared with hypertensive individuals without increased aldosterone production. Circulating aldosterone also produced a negative effect on various parameters of cardiac structure and function even in nonhypertensive subjects with GRA compared with age- and gender-matched control subjects.83 After matching for age, body mass index, cholesterol, triglycerides, and glucose levels, arterial wall stiffness was found to be increased in primary aldosteronism compared with essential hypertension.79 Hyperaldosteronism has been associated with widespread tissue fibrosis and increased remodeling of resistance vessels.74
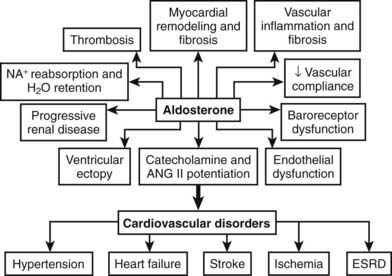
FIGURE 107-4. Schematic representation of the detrimental tissue actions of aldosterone leading to cardiovascular disorders. ESRD, End-stage renal disease.
Table 107-3. Cardiovascular Disease and Events in Primary Aldosteronism
Patients with primary aldosteronism have increased renal dysfunction compared with those with essential hypertension. In the relatively large Primary Aldosteronism Prevalence in Hypertensives (PAPY) Study from Italy, patients with APA or IHA had higher urinary microalbumin excretion compared with those with essential hypertension.80,81 Also, prevalence of the metabolic (insulin resistance) syndrome is increased in patients with primary aldosteronism compared with those with essential hypertension.82
Epidemiology of Primary Aldosteronism
Primary aldosteronism occurs in patients of all ages, including children. In children, growth failure may be the presenting feature. As in children with other causes of hypokalemia such as Bartter’s syndrome, growth failure may be the presenting feature. Patients with APA are usually younger than those with IHA.66,84 However, primary aldosteronism in children younger than 16 years is usually due to adrenal hyperplasia. Among reported children with adrenal adenoma causing primary aldosteronism, most are female.
At any age, adenomas are more common in females than in males.2,45 IHA has been found by some to be equally common in males and females46 and by others to be more common in males.84
Diagnosis of Primary Aldosteronism
General Considerations
In 2008, The Endocrine Society published a clinical guideline on the detection, diagnosis, and treatment of primary aldosteronism with specific recommendations for diagnostic workup.7 Sequential steps recommended for workup include (1) case detection (screening), (2) confirmation of the diagnosis, and (3) subtype classification.7
The approach to the diagnosis of primary aldosteronism differs from center to center. In some centers, the only patients investigated are those with hypokalemia and hypertension. As discussed above, most patients with this syndrome are normokalemic. Thus, a large fraction of patients with primary aldosteronism will not be detected if this approach is taken. In other centers, all hypertensive patients are screened for this diagnosis. Favoring this approach, the cost involved in screening is relatively small compared with the cost of lifelong drug therapy and the potential benefit derived from surgical cure or specific medical treatment.
Clinical criteria and methods for screening for primary aldosteronism are summarized in Fig. 107-5. All patients with the combination of hypertension and hypokalemia (spontaneous or diuretic induced) should be screened. Other recommendations for screening include young patients with hypertension, patients with early onset of cerebrovascular accident (<age 50), patients with a positive family history of early stroke in a first-degree relative, patients with hypertension and adrenal incidentalomas,85 those whose blood pressure is difficult to control (resistant hypertension), and those in whom the diagnosis of secondary hypertension is being entertained for other reasons.7
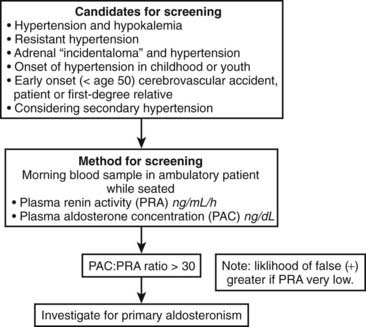
Primary hyperaldosteronism is especially common among patients with resistant hypertension. Resistant hypertension is defined as blood pressure that remains above goal despite concurrent use of three antihypertensive agents of different classes, or that requires a minimum of four agents to achieve therapeutic goal.86 In evaluations by several different investigators, the prevalence of primary hyperaldosteronism has been consistently reported to be about 20% among subjects with resistant hypertension.86 In support of hyperaldosteronism as a common cause of resistant hypertension, aldosterone antagonists have been shown to provide significant additional blood pressure reduction when added to existing multidrug regimens in patients whose blood pressure remains uncontrolled.86
At Mayo Clinic, an average of 12 patients per annum were diagnosed with primary aldosteronism from 1960 to 1991. However, with intensive screening, the number of patients with this diagnosis increased 10-fold over the subsequent 8 years.58 In the past, the tipoff for thinking of the disease was the measurement of serum potassium in a hypertensive patient. The presence of hypokalemia (serum potassium <3.6 mmol/L) in an untreated hypertensive patient demanded further investigation of the renin-angiotensin-aldosterone system. As was previously discussed, it is also worthwhile to investigate patients who have diuretic-induced hypokalemia.87 It is worth keeping in mind that circulating potassium levels may be elevated by red blood cell hemolysis and/or by the use of a tourniquet with muscle pumping.88 In an otherwise hypokalemic patient, normokalemia may be induced by low sodium intake because urinary potassium excretion is directly related to the distal nephron sodium load in conditions of mineralocorticoid excess.89,90 If 24-hour urinary sodium excretion is less than 100 mmol, hypokalemia may be provoked by increasing sodium intake (e.g., NaCl 6 g/day for 5 days) and then repeating the serum potassium measurement. Under these circumstances, some patients with normokalemic primary aldosteronism will become frankly hypokalemic. Twenty years ago, plasma or serum potassium was thought to have a sensitivity of 75% to 90% in the diagnosis of primary aldosteronism.15,91 Current data from centers employing aldosterone:PRA ratios for case detection suggest that this figure should be reduced to less than 50%. The precise prevalence of normokalemic primary aldosteronism in an untreated hypertensive population is currently unknown but now appears to be much higher than was previously appreciated. Bravo and associates92 reported 80 patients with primary aldosteronism, 27.5% of whom had normal serum potassium. In another study,66 11% of patients with primary aldosteronism were normokalemic. Many other laboratories have confirmed these findings.15,85 Thus, even before the aldosterone:renin ratio was introduced as the screening test of choice, between 7% and 38% of patients with primary aldosteronism had basal circulating potassium levels greater than 3.6 mmol/L. After screening with the ratio was introduced, 60% to 70% of patients with primary aldosteronism were reported as being normokalemic.
In general, hypokalemia secondary to mineralocorticoid excess is associated with inappropriate renal loss of potassium (kaliuresis). The degree of kaliuresis depends on daily potassium intake, but potassium excretion usually exceeds 30 mmol/24 hour in hypokalemic patients with primary aldosteronism (Fig. 107-6). In addition, enhanced distal nephron sodium-hydrogen exchange leads to increased hydrogen ion excretion, usually as ammonium ion, which is responsible for the relatively mild metabolic alkalosis observed in these patients. Although it is not understood, some patients with GRA have alkalosis but are persistently normokalemic.
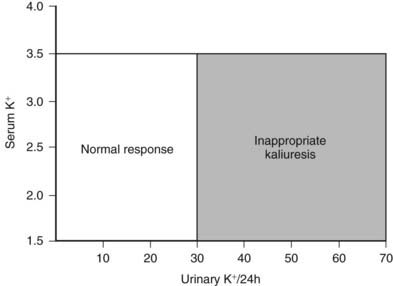
FIGURE 107-6. Relationship of urinary potassium (K+) excretion to serum K+ in normal subjects and patients with primary mineralocorticoid excess states, who have inappropriate kaliuresis.
In primary aldosteronism, plasma sodium levels usually are in the upper part of the normal range or are slightly elevated owing to resetting of the hypothalamic osmostat. As with other biochemical parameters, this abnormality may be more marked in patients with an adenoma than in those with hyperplasia.66 Total exchangeable sodium is increased in patients with primary aldosteronism caused by an adenoma but is usually normal in those with idiopathic adrenal hyperplasia.32 A similar situation exists for total exchangeable potassium, with reduced levels in patients with an adenoma but not in those with hyperplasia.32 Whether differences also occur between IHA and the angiotensin-responsive variety of adenoma (which biochemically mimics IHA in other ways, including responsiveness of aldosterone to angiotensin II and normalcy of “hybrid steroid” [18-hydroxy-cortisol and 18-oxo-cortisol] levels93) has yet to be determined.
Assessment of the Renin-Angiotensin-Aldosterone Axis
Plasma and Urinary Aldosterone
The fundamental abnormality of primary aldosteronism is excessive production of aldosterone, which is independent of the renin-angiotensin system and occurs in the face of suppressed PRA. Various factors, including the effects of antihypertensive drug therapy on the renin-angiotensin system and the inhibiting effect of hypokalemia directly on aldosterone secretion, may render the diagnosis difficult.
If serum potassium is <3.0 mmol/L, potassium supplementation should be administered and normokalemia established before aldosterone is measured.94,95 However, even when hypokalemia is corrected, plasma aldosterone levels may still sometimes be at the upper part of the normal range among patients with primary aldosteronism, even in those with APA. An acceptable alternative to plasma aldosterone measurement is the 24-hour urinary aldosterone excretion rate, which quantifies its acid-labile conjugate aldosterone-18-glucuronide.96 Although urinary aldosterone quantifies only approximately 15% of the aldosterone secreted, it provides a reliable index of aldosterone secretion in the absence of severe renal dysfunction. Assays to measure urinary tetrahydro-aldosterone (15% to 40% of metabolites) are employed less commonly. As with the measurement of plasma aldosterone, the urinary aldosterone excretion rate may be normal in primary aldosteronism, especially in the presence of significant hypokalemia.94,95 Other reasons for normal urinary aldosterone excretion in patients with primary aldosteronism include renal failure, incomplete collection of urine, and variability in rates of hepatic metabolism of aldosterone. Some patients, including some with apparently isolated 11-deoxycorticosterone (DOC) excess, have periodic aldosterone hypersecretion.97,98 Unlike plasma aldosterone, the urinary aldosterone excretion rate decreases with age.99 Therefore, it is important to use an age-related normal reference range. Also, for interpretation, a plasma aldosterone concentration (PAC) of 1 ng/dL translates into 27.7 pmol/L in Système International d’Unitès (SI units).
Plasma Renin Activity
The renin-angiotensin system usually is assessed by measuring PRA, which reflects the quantity of circulating active renin. This assay depends on the generation of angiotensin I in vitro in the patient’s plasma, which is quantified by radioimmunoassay. The validity of the assay as an index of renin secretion depends on the adequacy of renin substrate (angiotensinogen) in the patient’s plasma, but this is only rarely a problem. In primary aldosteronism, PRA is usually low or undetectable in contrast to the elevated levels found in secondary aldosteronism. However, PRA alone lacks specificity as a screening test for primary aldosteronism because it does not distinguish primary aldosteronism from low renin essential hypertension (LREH), which occurs in approximately 25% of patients with essential hypertension. Renin secretion is stimulated by assumption of erect posture or by sodium/volume depletion (as with diuretic treatment) and is suppressed in situations in which β-adrenergic input to the juxtaglomerular (JG) cells is abrogated (as in treatment with a β-adrenergic receptor blocker). Renin production also decreases during the aging process and in patients with chronic renal failure, owing to reduction in functioning JG cells and salt and water retention. In addition, renin secretion has a diurnal variation with highest levels in the morning. Therefore, to correctly interpret the test and PRA values, it is necessary to know the time of day and the patient’s age, posture, medical treatment, renal function, and, if possible, dietary sodium intake. Sodium intake can be estimated by concurrent measurement of 24-hour urinary sodium excretion rate and PRA. Also for interpretation, a PRA level of 1 ng/mL/hr in conventional units translates to 12.8 pmol/L/min in SI units.
Aldosterone-Renin Ratio
Introduced by Hiramatsu and colleagues100 in 1981, the aldosterone:renin ratio is recognized worldwide as the most reliable means of screening for primary aldosteronism.48,100–102 As indicated previously, this approach has uncovered a surprisingly large number of hypertensive patients with primary aldosteronism.
McKenna and colleagues102 found that a single elevated aldosterone:renin ratio associated with an elevated or normal plasma aldosterone value correctly diagnosed primary aldosteronism in 10 patients—5 with hyperplasia and 5 with APA. The only problem in this study with false-positive results involved patients with chronic renal failure.102 Secondary aldosteronism was characterized by elevated plasma aldosterone values together with a normal ratio.
In patients being considered for primary aldosteronism, screening can be accomplished by measuring a morning (8:00 to 10:00 am) random PAC and PRA (see Fig. 107-5). This test may be performed while the patient is taking antihypertensive agents (except for spironolactone and eplerenone) and does not require postural stimulation.22,103–106 As was mentioned earlier, hypokalemia reduces the secretion of aldosterone and should be corrected before testing is begun. Patients are encouraged to maintain a liberal dietary sodium intake while undergoing screening to prevent stimulation of renin (and a false-negative ratio) induced by dietary sodium restriction. Mineralocorticoid receptor antagonists spironolactone and eplerenone and high-dose amiloride are the only medications that absolutely interfere with interpretation of the aldosterone:renin ratio and should be discontinued for 5 to 6 weeks before testing is begun.106 β-Adrenoceptor blocking drugs, renal impairment, and old age can produce false-positive ratios as the result of renin suppression, whereas diuretics, ACE inhibitors, angiotensin receptor blockers, and dihydropyridine calcium channel blockers can produce false-negatives.
Antihypertensive agents that can be used to control blood pressure during a workup for primary aldosteronism are listed in Table 107-4. The α1-adrenoceptor blockers (prazosin, doxazosin, and terazosin), hydralazine, slow-release verapamil, and α-methyl-DOPA all have minimal effects on the aldosterone:renin ratio in patients with primary aldosteronism and can thus be employed. Slow-release verapamil (120 mg twice daily) is usually well tolerated (unless constipation develops). Side effects from hydralazine are rare if low doses (e.g., 12.5 mg twice daily) are given initially, increasing in a stepwise fashion (e.g., dose increments every 2 weeks) as required, and combination treatment with slow-release verapamil prevents the reflex tachycardia that can occur with “unopposed” use of a direct vasodilator, such as hydralazine. With the above agents, it is usually possible to control severe hypertension satisfactorily after withdrawal of other drug therapy during the process of diagnostic workup.7
Table 107-4. Clinical Considerations for Screening for Primary Aldosteronism Using the Plasma Aldosterone Concentration:Plasma Renin Activity Ratio (ARR)
Nonsteroidal antiinflammatory drugs, by promoting salt and water retention (suppressing renin) and hyperkalemia (stimulating aldosterone secretion), are frequently associated with false-positive ratios and should be withheld for several weeks before testing, if possible.
Interpretation of the PAC:PRA ratio for case detection purposes is relatively straightforward (see Table 107-3). Values ≥30 (when PAC is expressed as ng/dL and PRA as ng/mL/hr; conventional units); or equivalent to 750 when PAC is reported as pmol/L (SI units) and PRA in conventional units; or equivalent to 60 when PAC and PRA are expressed as SI units are generally accepted as positive for primary aldosteronism, with a range of 20 to 40 for most centers.7 It is important to point out that the lower limit of detection varies among the different PRA assays, and these variations can have a dramatic effect on the PAC:PRA ratio. Very low PRA values can increase the ratio when the PAC value is perfectly normal. To avoid this potential error, some groups have designated a minimal PAC value (≥15 ng/dL) necessary to interpret the ratio.7,58 Thus, when PAC is ≥15 ng/dL, then PAC:PRA ≥20 is considered positive.58 A high PAC:PRA ratio constitutes a positive screening test for primary aldosteronism, but it is important to realize that screening should be followed by an aldosterone suppression test to confirm the diagnosis prior to subtype classification.7,58
Establishment of the Diagnosis of Primary Aldosteronism
Instead of proceeding directly from a positive screening test to subtype classification, it is strongly recommended that patients undergo one of four aldosterone suppression tests (see below) to definitively confirm or exclude the diagnosis of primary aldosteronism (Table 107-5).7 Three of these are salt-loading tests that can be conducted by increasing dietary sodium intake, infusing saline, or administering exogenous mineralocorticoid, or through combinations of these approaches. The rationale behind these tests is that in normal subjects, volume expansion with saline suppresses plasma aldosterone, whereas in primary aldosteronism, further volume expansion does not have the same suppressive action on aldosterone secretion. The fourth test depends on inhibition of aldosterone secretion by an ACE inhibitor. Detailed methods for performing these tests have been reviewed recently.7 At present, evidence is insufficient to allow recommendation of any one of these tests over any of the others.7 It has been recommended that pharmacologic agents with minimal or no effect on the renin-angiotensin-aldosterone system (see Table 107-4; discussed above) be employed to control blood pressure during confirmatory testing, if possible. Whether or not medications that affect the renin-angiotensin-aldosterone system are discontinued, however, spironolactone and eplerenone clearly interfere with these tests and must be discontinued 5 to 6 weeks before testing in all cases.
Table 107-5. Comparison of the Four Accepted Confirmation Tests for the Diagnosis of Primary Aldosteronism With Normal and Abnormal Values
IV, Intravenous; Na+, sodium; PAC, plasma aldosterone concentration; TID, three times daily.
Fludrocortisone Suppression Test
In 1967, Biglieri and associates97 demonstrated that 11-deoxycorticosterone (DOC) acetate administration for 3 days failed to suppress urinary aldosterone excretion in APA but suppressed aldosterone secretion both in normal subjects and in two patients with primary aldosteronism in whom no tumor was found at surgery (probable idiopathic adrenal hyperplasia). This observation suggested that exogenous mineralocorticoid administration might be of value in the diagnosis and differential diagnosis of primary aldosteronism. Subsequent studies, however, reported that some patients with IHA also failed to suppress aldosterone secretion.15,107 Extracellular fluid volume expansion with a combination of a high-sodium diet and oral fludrocortisone, over a 4-day period, is now regarded as a reliable means of definitively confirming or excluding the diagnosis of primary aldosteronism.108,109 The reliability of the test is dependent upon the maintenance of normokalemia by potassium chloride supplementation (otherwise, hypokalemia may result in a fall in aldosterone concentrations and a false-negative test) and the demonstration of adequate suppression of PRA (to <1 ng/mL/hr). Additional details regarding the fludrocortisone suppression test have been published.7,110,111 The Brisbane group has established that PAC >6 ng/dL at 10 am on day 4 of salt loading and fludrocortisone confirms the diagnosis if PRA is <1 ng/mL/hr.111 Some centers conduct this test in the ambulatory clinic; others require a few days of hospitalization.
Oral Sodium Loading Test
Failure of suppression of the urinary aldosterone excretion rate in patients on a high-sodium diet (>200 mEq/d) is a valid confirmatory test of primary aldosteronism.58,92,112 After 3 days on high oral sodium intake, a normal individual is expected to suppress aldosterone excretion to <12 µg/24 hr. In contrast, patients with primary aldosteronism do not suppress urinary aldosterone excretion rates to <12 µg/24 hr, provided that serum potassium concentration is maintained in the normal range by potassium chloride supplementation during the salt-loading period.58,112
Intravenous Saline Suppression Test
Another valid confirmatory test is to measure plasma aldosterone concentration after intravenous saline infusion (2 L normal saline over 4 hr).113 This approach has been used successfully to discriminate between patients with essential hypertension and those with primary aldosteronism. Patients with essential hypertension demonstrate suppression of plasma aldosterone to <5 ng/dL; those with primary aldosteronism do not suppress plasma aldosterone to <10 ng/dL; those with aldosterone suppression to 5 to 10 ng/dL fall into the indeterminate range and may require retesting at a later time. The protocol for this test is to measure basal plasma aldosterone with the patient in the recumbent position at 8:00 am. Subsequently, after 2 L of normal saline is infused over 4 hours, plasma aldosterone concentration is measured again. None of the above salt-loading tests (with or without exogenous mineralocorticoid) should be performed in patients with severe uncontrolled hypertension, severe renal insufficiency, heart failure, cardiac arrhythmia, or severe hypokalemia.7
Captopril Challenge Test
In normal subjects on normal sodium intake, oral administration of an ACE inhibitor (e.g., captopril) reduces plasma aldosterone levels. This reduction does not occur in patients with primary aldosteronism. The usual test involves giving captopril 25 mg orally and measuring plasma aldosterone levels 2 hours later. In patients with primary aldosteronism, the 2 hour level remains >15 ng/dL in contrast to the decrease in normal subjects.114 Not all investigators have found this approach valuable.115,116 Muratani and colleagues116 studied 19 patients with primary aldosteronism and 72 with essential hypertension. Captopril was administered after overnight recumbency. The test was 93% specific and had a 79% predictive value. However, higher specificity (97%) and predictive values (90%) were obtained via analysis of the pre-captopril plasma aldosterone:PRA ratios.
The effect of dietary sodium intake on the captopril challenge test was investigated by Naomi and coworkers.117 The authors used a higher dose of captopril (50 mg) and found that results of the test were unaffected by altering the sodium intake. They analyzed the PAC:PRA ratios in blood samples taken 90 minutes after oral captopril was given at 9:00 am and after 1 hour of recumbency. Using a ratio greater than 20 for the diagnosis of primary aldosteronism, they found that the test had 95% sensitivity and 92% specificity. Numerous cases of false-negative or equivocal results were reported with the captopril challenge test.7
Dexamethasone Suppression Test
Patients with GRA respond to confirmatory tests of the renin-angiotensin-aldosterone system in a manner similar to that of patients with APA. These two conditions can be distinguished, however, by the patient’s response to exogenous glucocorticoid administration. With the patient in the upright position in the morning, plasma aldosterone concentration below 5 ng/dL after overnight dexamethasone administration (1.0 mg at midnight and 0.5 mg at 6:00 am) has been reported as a cutoff point to separate patients with GRA from those with IHA or APA.118 The distinction between GRA and other forms of primary aldosteronism becomes clearer with long-term glucocorticoid therapy. Long-term dexamethasone (e.g., 2 mg/d for 3 weeks) in GRA (but not in other forms of primary aldosteronism) leads to recovery of the suppressed renin-angiotensin system, normalization of plasma potassium and aldosterone, reduction of blood pressure (usually to normal), and restoration of responsiveness of the zona glomerulosa to angiotensin II.119–121
The need for dexamethasone suppression testing in GRA has now been largely supplanted by the introduction of genetic testing, which is 100% sensitive and specific.28,121
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


