FIGURE 109-1. A, Anatomic distribution of extra-adrenal chromaffin tissue in the newborn. B, Locations of extra-adrenal pheochromocytomas reported in the literature up to 1965.
(From Coupland R: The Natural History of the Chromaffin Cell, Essex, UK, Longsman Green, 1965.)
Most pheochromocytomas arise sporadically, but based on recent reports, up to 24% are familial.12 In contrast to sporadic pheochromocytomas that usually are unicentric and unilateral, familial pheochromocytomas are often multifocal and bilateral.1,5,13
History
The name pheochromocytoma, proposed by Pick in 1912,14 comes from the Greek words phaios (“dusky”) and chroma (“color”); it refers to the staining that occurs when the tumors are treated with chromium salts. The first diagnosis of pheochromocytoma was made in 1886 by Frankel,15 who found bilateral tumors of the adrenal gland at autopsy in an 18-year-old girl who had died suddenly. Extra-adrenal pheochromocytoma was first reported by Alezais and Peyron in 1908.16 The important association of paroxysmal hypertension with pheochromocytoma was first described by L’Abbe and colleagues in 1922.17 The first successful surgical removals of pheochromocytomas were performed by Roux in France in 1926 and by C. H. Mayo in the United States in 1927.18,19 In 1929, Rabin20 reported that a pheochromocytoma contained an excess amount of the normal pressor agent present in the adrenal gland, and suggested that this might account for the main clinical manifestations (i.e., hypertension and tachycardia). In 1936, epinephrine (also known as adrenaline) was isolated from a pheochromocytoma by Kelly and colleagues,21 but it was not until 1946 that von Euler and his coworkers, and 1947 that Holtz and his coworkers, reported independently the occurrence of norepinephrine (noradrenaline) in the body.22–24 In 1949, Holton25 first demonstrated the presence of norepinephrine in a pheochromocytoma. The first large series of successful surgical removals with pharmacologic blockade was reported by Kvale and colleagues in 1956.26
According to different reviews and statistics, pheochromocytomas account for approximately 0.05% to 0.1% of patients with any degree of sustained hypertension1,5,18; however, this probably includes only 50% of persons harboring the tumor, when it is considered that about half the patients with pheochromocytoma have only paroxysmal hypertension or are normotensive. Also, despite the low incidence of pheochromocytoma in patients with sustained hypertension, it must be considered that the current prevalence of sustained hypertension in the adult population of Western countries is up to 30%.27–29 In Western countries, the prevalence of pheochromocytoma can be estimated at between 1 : 4500 and 1 : 1700, with an annual incidence of 3 to 8 cases per 1 million per year in the general population.30 Pheochromocytoma occurs at any age, but most often in the fourth and fifth decades, and it occurs equally in men and women.
Catecholamine Synthesis, Release, and Metabolism
Pheochromocytomas are often described as tumors that synthesize, store, and secrete catecholamines. It is rarely appreciated that pheochromocytomas also metabolize catecholamines, and that this is a more consistent process than that of catecholamine secretion.31 Failure to recognize this key feature is probably caused by several misconceptions about the storage and metabolism of catecholamines, perhaps the most pervasive being that metabolism occurs mainly after catecholamine release. In fact, substantial amounts of catecholamines are metabolized in the same cells where the amines are synthesized, and much of this is independent of catecholamine release. The nature of the metabolites produced depends on the neuronal or chromaffin cell phenotype and the types of catecholamines synthesized within the tumor cells.
BIOSYNTHESIS
Catecholamine synthesis begins with the amino acid tyrosine, which comes from the diet or via hydroxylation of phenylalanine in the liver. l-Tyrosine is converted to dihydroxyphenylalanine (dopa) by tyrosine hydroxylase (TH) (Fig. 109-2). This reaction requires molecular oxygen and the cofactor tetrahydrobiopterin and represents the rate-limiting step in catecholamine biosynthesis (Fig. 109-3).32 Tissue sources of catecholamines therefore are principally dependent on the presence of this enzyme, which is largely confined to dopaminergic and noradrenergic neurons of the central nervous system, and to sympathetic nerves and chromaffin cells of the adrenal medulla and extramedullary paraganglia in the periphery.
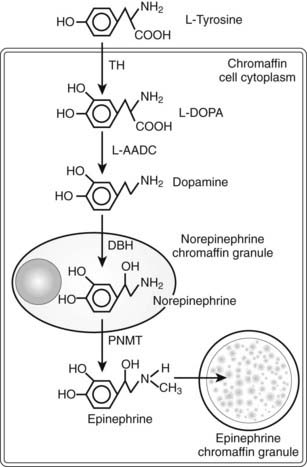
FIGURE 109-2. Diagram illustrating the catecholamine biosynthetic pathway in an adrenal chromaffin cell. DBH, Dopamine β-hydroxylase; L-AADC, L-aromatic amino acid decarboxylase; PNMT, phenylethanolamine N-methyltransferase; TH, tyrosine hydroxylase.
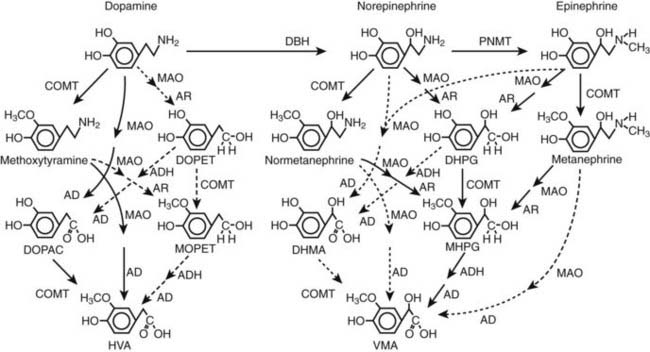
FIGURE 109-3. Pathways of metabolism of catecholamines. Enzymes responsible for each pathway are shown at the heads of arrows. Solid arrows indicate the major pathways, whereas dotted arrows indicate pathways of negligible importance. Pathways of sulfate conjugation are not shown. AD, Aldehyde dehydrogenase; ADH, alcohol dehydrogenase; AR, aldose or aldehyde reductase; COMT, catechol O-methyltransferase; DBH, dopamine β-hydroxylase; DHMA, 3,4-dihydroxymandelic acid; DHPG, 3,4-dihydroxyphenylglycol; DOPAC, 3,4-dihydroxyphenylacetic acid; DOPET, 3,4 dihydroxyphenylethanol; HVA, homovanillic acid; MAO, monoamine oxidase; MHPG, 3-methoxy-4 hydroxyphenylglycol; MOPET, 3-methoxy-4 hydroxyphenyletanol; PNMT, phenolethanolamine N-methyltransferase; VMA, vanillylmandelic acid.
Dopa is decarboxylated by aromatic l-amino acid decarboxylase to produce dopamine. The dopamine formed in neurons and chromaffin cells is translocated from the cytoplasm into vesicular storage granules. In dopaminergic neurons of the central nervous system, dopamine serves as the end product neurotransmitter. Large amounts of dopamine are produced as an end product of catecholamine synthesis in peripheral non-neuronal cells of the gastrointestinal tract and kidneys.33 The dopamine present in urine is derived mainly from decarboxylation of dopa taken up from the circulation by the kidneys.
The dopamine formed in noradrenergic neurons and chromaffin cells is converted to norepinephrine by dopamine β-hydroxylase (DBH), an enzyme that is found only in the vesicles of cells that synthesize norepinephrine and epinephrine.
In adrenal medullary chromaffin cells, norepinephrine is metabolized by the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT) to form epinephrine.34 Epinephrine then is translocated into chromaffin granules, where the amine is stored while awaiting release.35 PNMT activity is induced by high levels of glucocorticoid production in the adrenal cortex.
Presumably because expression of PNMT is dependent on high local concentrations of glucocorticoids, pheochromocytomas that produce large amounts of epinephrine typically have an adrenal location, whereas those in extra-adrenal locations usually produce only norepinephrine.36 Pheochromocytomas that lack both PNMT and DBH and that predominantly produce dopamine are extremely rare. Dopamine production is a more common finding in individuals with parasympathetic-associated paragangliomas.
Conversion of tyrosine to dopa by TH represents the pivotal step in regulating synthesis and in maintaining stores of catecholamines in response to changes in catecholamine turnover associated with variations in exocytotic release. Rapid activation of TH is achieved by phosphorylation of serine residues at the regulatory domain, under the control of multiple Ca2+ and cyclic adenosine monophosphate (cAMP)-dependent pathways influenced by changes in nerve activity and actions of peptides and other coactivators.37 Long-term regulation involves induction of synthesis of the enzyme at the transcriptional level. Feedback inhibition by catecholamines provides a further mechanism for the short-term regulation of enzyme activity.
α-Methyl-l-tyrosine or metyrosine (Demser) is an analogue of tyrosine that inhibits TH, thereby decreasing catecholamine stores. The drug is used occasionally in patients with pheochromocytoma (particularly those with extensive metastatic disease) or preoperatively.
STORAGE AND RELEASE
Translocation of catecholamines into vesicular granules for storage is facilitated by two vesicular monoamine transporters.38 Both transporters have wide specificity for different monoamine substrates. It is important to note that vesicular stores of catecholamines do not exist in a static state, simply waiting for a signal for exocytotic release. Rather, vesicular stores of catecholamines exist in a highly dynamic equilibrium with the surrounding cytoplasm, with passive outward leakage of catecholamines into the cytoplasm counterbalanced by inward active transport under control of the vesicular monoamine transporters.31
Catecholamines share the acidic environment of the storage granule matrix with adenosine triphosphate (ATP), peptides, and proteins, the most well known of which are the chromogranins.39 The chromogranins are ubiquitous components of secretory vesicles, and their widespread presence among endocrine tissues has led to their measurement in plasma as useful, albeit relatively nonspecific, markers of neuroendocrine tumors, including pheochromocytomas.
Catecholamines are stored in several types of vesicular granules that differ in size and type of protein and peptide components, the specific functions of which are incompletely understood. Two populations of chromaffin cells have been described with morphologically distinct vesicles, which preferentially store norepinephrine or epinephrine, and which release the two catecholamines differentially in response to different stimuli. Most adrenal pheochromocytomas secrete both norepinephrine and epinephrine; about a third exclusively produce norepinephrine, and a much smaller proportion exclusively produce and secrete epinephrine.
The process of exocytosis occurs at specialized locations on sympathetic varicosities dictated by the cell-surface expression of specialized docking proteins that interact with other proteins on the surfaces of secretory vesicles. The process is stimulated by an influx of Ca2+, which in neurons is controlled primarily by nerve impulse–mediated membrane depolarization, and in adrenal medullary cells by acetylcholine release from innervating splanchnic nerves. The wide range of voltage-, receptor-, G protein–, and second messenger–operated Ca2+ channels provide numerous points for regulation of Ca2+-triggered exocytosis. In this way, a variety of peptides, neurotransmitters, and humoral factors provide additional mechanisms for stimulation of exocytosis or may act to modulate nerve impulse–stimulated release of catecholamines. Norepinephrine also inhibits its own release through occupation of presynaptic α2-adrenoceptors. The mechanisms regulating catecholamine release are closely coordinated so as to regulate the enzymes responsible for synthesis, thereby ensuring appropriate replenishment of the catecholamines lost through exocytosis.40
UPTAKE AND METABOLISM
Because the enzymes responsible for the metabolism of catecholamines have intracellular locations, the primary mechanism limiting the life span of catecholamines in the extracellular space is uptake by active transport, not metabolism by enzymes.41 Uptake is facilitated by transporters that belong to two families with mainly neuronal and extraneuronal locations. The neuronal norepinephrine transporter provides the principal mechanism for rapid termination of the signal in sympathoneuronal transmission, whereas transporters at extraneuronal locations are more important for limiting the spread of the signal and for clearing catecholamines from the bloodstream. For norepinephrine released by sympathetic nerves, about 90% is removed back into nerves by neuronal uptake, 5% is removed by extraneuronal uptake, and 5% escapes these processes to enter the bloodstream. In contrast, for epinephrine released directly into the bloodstream from the adrenals, about 90% is removed by extraneuronal monoamine transport processes, the liver being particularly important. The presence of these highly active transport processes means that catecholamines are rapidly cleared from the bloodstream with a circulatory half-life of less than 2 minutes.
Irreversible inactivation of released catecholamines occurs in a series arrangement, with uptake followed by metabolism. Metabolism occurs through a multiplicity of pathways catalyzed by an array of enzymes and resulting in a variety of metabolites (see Figs. 109-3 and 109-4).42 Deamination of catecholamines by monoamine oxidase (MAO) yields reactive aldehyde intermediate metabolites that are metabolized further to deaminated acids (by aldehyde dehydrogenase) or to deaminated alcohols (by aldehyde or aldose reductase). The aldehyde intermediate formed from dopamine is a good substrate for aldehyde dehydrogenase but not for aldehyde or aldose reductase. In contrast, the aldehyde intermediates formed from the β-hydroxylated catecholamines (i.e., norepinephrine and epinephrine) are good substrates for aldehyde or aldose reductase but are poor substrates for aldehyde dehydrogenase. Therefore, norepinephrine and epinephrine are preferentially deaminated to 3,4-dihydroxyphenylglycol (DHPG), the alcohol metabolite. Deamination to the deaminated acid metabolite, 3,4-dihydroxymandelic acid (DHMA), is not a favored pathway.
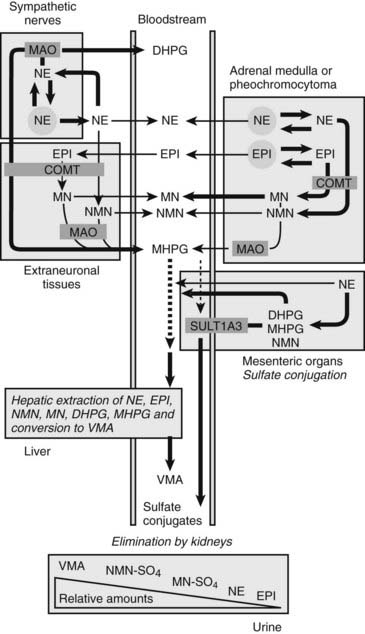
FIGURE 109-4. Model showing contributions of the adrenal medulla or a pheochromocytoma, in relation to sympathetic nerves and other tissues, to production of circulating catecholamines and catecholamine metabolites. Most metabolism occurs in the same cells where catecholamines are synthesized, mainly from catecholamines leaking from storage granules. In sympathetic nerves, where normally most catecholamine metabolism occurs, norepinephrine (NE) is deaminated by monoamine oxidase (MAO) to 3,4-dihydroxyphenylglycol (DHPG). The additional presence of catechol O-methyltransferase (COMT) in adrenal medullary cells leads to conversion of NE to normetanephrine (NMN) and of epinephrine (EPI) to metanephrine (MN). Normally, O-methylation is a minor pathway of catecholamine metabolism, but when a pheochromocytoma is present, O-methylation can represent a major pathway of metabolism. NMN and MN are converted to the sulfate-conjugated metabolites by sulfotransferase type 1A3 (SULT1A3), an enzyme present in high concentrations in digestive tissues. Vanillylmandelic acid (VMA), the main catecholamine metabolite excreted in urine, is formed in the liver largely from methoxyhydroxyphenylglycol (MHPG). MHPG is normally largely derived from extraneuronal O-methylation of DHPG produced in sympathetic nerves.
Catechol-O-methyltransferase (COMT) is responsible for the second major pathway of catecholamine metabolism: catalyzing O-methylation of dopamine to methoxytyramine, norepinephrine to normetanephrine, and epinephrine to metanephrine. COMT is not present in catecholamine-producing neurons, which contain exclusively MAO, but it is present along with MAO in most extraneuronal tissues. The membrane-bound isoform of COMT, which has high affinity for catecholamines, is especially abundant in adrenal chromaffin and pheochromocytoma tumor cells.43 As a result of differences in expression of metabolizing enzymes, catecholamines produced in neurons and adrenal medullary or pheochromocytoma tumor cells follow different neuronal and extraneuronal pathways of metabolism (see Figs. 109-3 and 109-4).
Neuronal pathways are quantitatively far more important than are extraneuronal pathways for the metabolism of norepinephrine produced in sympathetic nerves (see Fig. 109-4). The reasons for this are twofold. First, much more norepinephrine released by sympathetic nerves is removed by neuronal uptake than by extraneuronal uptake. Second, under resting conditions, much more of the norepinephrine metabolized intraneuronally is derived from transmitter leaking from storage vesicles than from transmitter recaptured after exocytotic release. Thus, most of the norepinephrine produced in the body is metabolized initially to DHPG, mainly from transmitter deaminated intraneuronally after leakage from storage vesicles or after release and reuptake.
DHPG is further O-methylated by COMT in non-neuronal tissues to 3-methoxy-4-hydroxyphenylglycol (MHPG), a metabolite that is also produced to a limited extent by deamination of normetanephrine and metanephrine. Compared with DHPG, normetanephrine and metanephrine are produced in small amounts and only at extraneuronal locations, with the single largest source representing adrenal chromaffin cells, which account for more than 90% of circulating metanephrine and 24% to 40% of circulating normetanephrine.44 Within the adrenals, normetanephrine and metanephrine are produced similarly to DHPG in sympathetic nerves, from norepinephrine and epinephrine leaking from storage granules into the chromaffin cell cytoplasm.
The MHPG produced from DHPG and metanephrines is conjugated or metabolized to vanillylmandelic acid (VMA) by the sequential actions of alcohol dehydrogenase and aldehyde dehydrogenase. The former enzyme is localized largely to the liver. Thus, at least 90% of the VMA formed in the body is produced in the liver, mainly from hepatic uptake and metabolism of circulating DHPG and MHPG.45
With the exception of VMA, all the catecholamines and their metabolites are metabolized to sulfate conjugates by a specific sulfotransferase isoenzyme type 1A3 (SULT1A3). In humans, a single amino acid substitution confers the enzyme with particularly high affinity for dopamine and the O-methylated metabolites of catecholamines, including normetanephrine, metanephrine, and methoxytyramine. The SULT1A3 isoenzyme is found in high concentrations in gastrointestinal tissues, which therefore represent a major source of sulfate conjugates.
In humans, VMA and the sulfate and glucuronide conjugates of MHPG represent the main end products of norepinephrine and epinephrine metabolism. HVA and the conjugates of HVA are the main metabolic end products of dopamine metabolism. These end products and the other conjugates are eliminated mainly by urinary excretion. As a result, their circulatory clearance is slow and their plasma concentrations are high relative to those of the precursor amines.
Clinical Presentation
SIGNS AND SYMPTOMS
The presence of pheochromocytoma usually is characterized by clinical signs and symptoms (Table 109-1) that result from hemodynamic and metabolic actions of circulating catecholamines or, less commonly, of other amines and co-secreted neuropeptides.18 Clinical findings such as hypertension, headache, unusual sweating, frequent arrhythmias, and the presence of pallor during hypertensive episodes are highly suggestive of pheochromocytoma. Although headache, palpitations, and sweating are nonspecific symptoms, their presence in patients with hypertension should arouse immediate suspicion for a pheochromocytoma because this triad constitutes the most commonly encountered symptoms in patients with a pheochromocytoma.
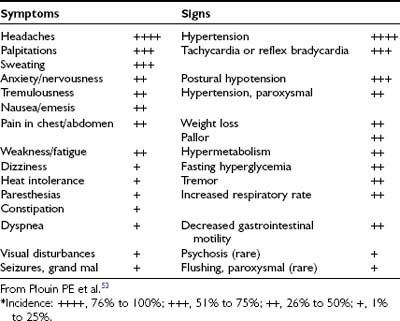
Sustained or paroxysmal hypertension (equally present) is the most common clinical sign (85% to 90%). Up to 13% of patients typically present with persistently normal blood pressure,5,18 but this proportion can be much higher in patients with adrenal incidentalomas or in those who undergo periodic screening for familial pheochromocytoma.12,46,47 In the latter group, tumors now are being found at an earlier stage, usually are quite small, and secrete low amounts of catecholamines. Other factors that may affect pheochromocytoma-associated elevations in blood pressure include the nature of catecholamine secretion, that is, receptor downregulation caused by consistently high levels of catecholamines, hypovolemia, and associated changes in sympathetic nerve function.48 Pheochromocytoma also may present with hypotension, particularly with postural hypotension or with alternating episodes of high and low blood pressure.5,49 This is commonly seen in patients who harbor tumors that are secreting epinephrine or compounds causing vasodilatation, or after higher doses of antihypertensive therapy. Hypotension also may occur secondary to hypovolemia, abnormal autonomic reflexes, or differential stimulation of α- and β-adrenergic receptors, or as a result of the type of neuropeptide that is co-secreted.
Headache, which occurs in up to 90% of patients with pheochromocytoma,1,18 may be mild or severe, short or long in duration, and may last for up to several days. In some patients, catecholamine-induced headache may be similar to tension headache.
Excessive generalized sweating occurs in approximately 60% to 70% of patients presenting with pheochromocytoma.1,18 Other complaints are palpitations and dyspnea, weight loss despite normal appetite (caused by catecholamine-induced glycogenolysis and lipolysis), and generalized weakness.49,50
Some patients present with new and commonly more severe episodes of anxiety or panic attacks.49 Palpitations, anxiety, and nervousness are more common in patients with pheochromocytomas that produce epinephrine.49 Less common clinical manifestations include fever of unknown origin (hypermetabolic state) and constipation secondary to catecholamine-induced decrease in intestinal motility, or possibly enkephalins.51 Some patients say that they feel flushed18,52; however, in our experience, the observation of an actual flush during a paroxysmal hypertensive episode in a patient with pheochromocytoma is an exceedingly rare event. Occasionally, Raynaud’s phenomenon is noted. Patients also may present with tremor, seizures, hyperglycemia, hypermetabolism, weight loss (usually noted only in patients with malignant pheochromocytoma), fever, and even mental changes.18,52
Pheochromocytoma-induced metabolic or hemodynamic attacks may last from a few seconds to several hours, with intervals between attacks varying widely; some occur only once every few months. A typical paroxysm is characterized by a sudden major increase in blood pressure; a severe, often pounding headache; profuse sweating over most of the body, especially the trunk; palpitations with or without tachycardia; prominent anxiety or a sense of doom; skin pallor; nausea, with or without emesis; and pain in the abdomen, the chest, or both.18,52,53 After an episode, patients usually feel drained and exhausted, and some may urinate more frequently. Initially, the episodes may be mild, of short duration, and infrequent.
Unusual symptoms related to paroxysmal blood pressure elevations during diagnostic procedures such as endoscopy, administration of anesthesia (caused by a sudden fall in blood pressure or any activation of the sympathetic nervous system, such as occurs during the induction phase of general anesthesia), or ingestion of food or beverages containing tyramine (e.g., certain cheeses, beers, wines, bananas, and chocolate) should arouse immediate suspicion of pheochromocytoma. The use of certain drugs such as histamine, metoclopramide, adrenocorticotropic hormone (ACTH), phenothiazine, methyldopa, monoamine oxidase inhibitors, tricyclic antidepressants, opiates (e.g., morphine, fentanyl), naloxone, droperidol, metoclopramide, glucagon, and chemotherapy may precipitate a hypertensive episode.18,52,54–60 Moreover, micturition or bladder distention in the case of a pheochromocytoma of the urinary bladder (more than half of these individuals have painless hematuria) should promptly arouse suspicion of the presence of this tumor. Causes that account for episodic catecholamine secretion often remain unestablished, but in some situations, it may be caused by intentional or accidental tumor manipulation coupled with an increase in intra-abdominal pressure from palpation, defecation, a fall, an automobile accident, or pregnancy.18,52,61 Psychological stress does not usually precipitate a hypertensive crisis.18,52,61 Timing of attacks may be unpredictable, and attacks may also occur at rest. However, about 8% to 10% of patients may be completely asymptomatic, usually because of a very small (less than 1 cm) tumor associated with nonsignificant catecholamine secretion or tumor dedifferentiation characterized by the absence of catecholamine-synthesizing enzymes and resulting in no production of catecholamines.5 Normal plasma and urine catecholamines and metanephrines due to absence of catecholamine production have been observed in patients with metastatic pheochromocytoma, often due to an underlying mutation of the succinate dehydrogenase subunit B gene.62 The defect was identified as an absence of tyrosine hydroxylase, the enzyme that catalyzes the initial and rate-limiting step in catecholamine biosynthesis (personal communication). These patients with so-called biochemically silent pheochromocytoma usually present at an advanced stage of the disease with symptoms and signs related to tumor mass effects rather than with symptoms and signs of catecholamine excess.
The hyperglycemia is usually mild, occurs with the hypertensive episodes, is accompanied by a subnormal level of plasma insulin (because of α-adrenergic inhibition of insulin release),63 and usually does not require treatment; however, it can be sustained and severe enough to require insulin and even to present as diabetic ketoacidosis.64 Hypoglycemia has also been reported.
Hypercalcemia has been reported in some patients with pheochromocytoma, sometimes as part of multiple endocrine neoplasia type 2 (MEN2)54 but at other times without evidence of parathyroid disease65 because the hypercalcemia disappears after the tumor is resected. Likewise, high levels of serum calcitonin, suggestive of medullary thyroid carcinoma, have been reported; these return to normal after the tumor is removed.66 In addition, pheochromocytoma has presented as Cushing’s syndrome with the tumor as the ectopic source of ACTH.67 Because catecholamines suppress intestinal motility, constipation is common, and occasionally even adynamic ileus occurs. Rarely, pheochromocytoma has produced vasoactive intestinal peptide with resultant watery diarrhea, hypokalemia, and achlorhydria (Verner-Morrison syndrome).68 Lactic acidosis without shock or sepsis has been reported.69 Interleukin-6 (IL-6) may have caused fever and multiorgan failure in one patient with pheochromocytoma with increased serum IL-6 and in another with pheochromocytoma and Castleman’s disease (IL-6–mediated B cell proliferation) because these conditions were reversed in each patient when the tumor was removed.70,71 Many patients with pheochromocytoma are asymptomatic or have only minor signs and symptoms; therefore, the diagnosis is easily missed, often with tragic consequences. Several studies of routine autopsies have indicated that most pheochromocytomas are first discovered after death.4 This pattern has continued into more recent studies, which show that the diagnosis is missed much more often in elderly patients.72 This may result from the fact that elderly patients often have other, more common diseases (e.g., coronary or cerebral atherosclerosis) that could easily explain the minor symptoms that patients report.72 A list of emergency situations characteristic for the presence of pheochromocytoma are described in Table 109-2.
Table 109-2. Emergency Situations Related to Catecholamine Excess Released from Pheochromocytoma
| Clinical Setting | Symptoms |
|---|---|
| Pheochromocytoma multisystem crisis (PMC) | Hypertension and/or hypotension, multiple organ failure, temperature of 40° C, encephalopathy |
| Cardiovascular | |
| Pulmonary | |
| Abdominal | |
| Neurologic | |
| Renal | |
| Metabolic |
Adapted from Brouwers FM, Lenders JW, Eisenhofer G, Pacak K: Pheochromocytoma as an endocrine emergency, Rev Endocr Metab Disord 4:121–128, 2003.
Estrogen, growth hormone, vitamin D, and retinol A (Accutane) have been shown to induce pheochromocytoma in experimental animals.73 Whether these hormones contribute to the higher incidence of clinical pheochromocytoma or to an increase in malignant potential is unknown; however, recently at least two patients chronically receiving Accutane treatment were found to have pheochromocytoma (personal communications).
In summary, the following patients should be evaluated for a pheochromocytoma: (1) anyone with the triad of headaches, sweating, and tachycardia, whether or not the subject has hypertension; (2) anyone with a known mutation of one of the susceptibility genes and/or a family history of pheochromocytoma; (3) anyone with an incidental adrenal mass; (4) anyone whose hypertension is associated with borderline increases in catecholamine production reflected by elevated plasma catecholamine or metanephrine levels; (5) anyone whose blood pressure is poorly responsive to standard therapy; and (6) anyone who has had hypertension, tachycardia, or an arrhythmia in response to anesthesia, surgery, or medications known to precipitate symptoms in patients with pheochromocytoma.48
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of pheochromocytoma includes a long list of conditions that may suggest the presence of the tumor (Table 109-3).18,52,61 Many of these conditions can be excluded readily on the basis of a good history and physical examination. The most common mimic is hyperadrenergic hypertension, which is characterized by tachycardia, sweating, anxiety, and increased cardiac output.74,75 These patients often have increased levels of catecholamines in blood and urine and may be excluded by use of the clonidine suppression test, which shows that the excess catecholamines are caused by excess central sympathetic nervous activity and are not caused by a tumor (see the following).48 Another common problem involves differentiating patients with pheochromocytoma from those with anxiety or panic attacks. This generally requires close observation of the patient during an episode.
Table 109-3. Differential Diagnosis for Pheochromocytoma
Ingestion of tyramine-containing foods or proprietary cold preparations while taking monoamine oxidase inhibitors |
Differentiation from an acute myocardial infarction may be difficult because angina pectoris and myocardial damage may occur in the absence of coronary artery disease in a patient with a pheochromocytoma. Many nonspecific electrocardiographic (ECG) changes have been reported, as well as various supraventricular and ventricular tachycardias, in patients with pheochromocytoma.
In the very rare situation in which a strong discordance is noted between symptoms and laboratory findings, one must consider the possibility of factitious administration of catecholamines. In this rare situation, as in other cases of factitious illness, an association of the individual with someone in the medical profession appears to be one of the common elements and should raise appropriate suspicion.
Patterns of biochemical test results that are more suggestive of sympathoadrenomedullary activation than of a tumor (e.g., as occurs in hypernoradrenergic hypertension, renovascular hypertension, congestive heart failure, panic disorder, and dumping syndrome) include proportionally larger elevations above the normal limits of plasma norepinephrine or epinephrine than of plasma normetanephrine or metanephrine.
Although severe paroxysmal hypertension should always raise suspicion of pheochromocytoma, it can also reflect a clinical entity called pseudopheochromocytoma. Pseudopheochromocytoma refers to the large majority of individuals (often women) with severe paroxysmal hypertension, whether normotensive or hypertensive between episodes, in whom pheochromocytoma has been ruled out.76,77 Recent evidence indicates that pseudopheochromocytoma is a heterogeneous clinical condition subdivided into a primary and a secondary form. In contrast to a primary form, a secondary form is associated with various pathologies (e.g., hypoglycemia, autonomic epilepsy, baroreceptor failure) and medications, or with drug abuse.
The most common clinical characteristics of this syndrome might be attributable in many cases to short-term activation of the sympathetic nervous system. Paroxysmal hypertension usually is associated with tachycardia, palpitations, nervousness, tremor, weakness, excessive sweating, pounding headache, feeling hot, and facial paleness or (rarely) redness. In contrast to pheochromocytoma, patients with pseudopheochromocytoma more often present with panic attacks or anxiety, flushing, nausea, and polyuria.76–78 All these symptoms well resemble a syndrome described by Page.79 Equally interesting were his observations that “an attack is brought on by excitement” and that the syndrome has a clear predominance in women. Among the important features distinctive from pheochromocytoma are the circumstances under which episodes occur. In pseudopheochromocytoma, symptoms may be provoked rarely by some identifiable event. It is important, therefore, in questioning these patients, to search for specific provocative factors that may have precipitated these episodes. Similar to pheochromocytoma, episodes may last from a few minutes to several hours and may occur daily or once every few months. Between episodes, blood pressure is normal or may be mildly elevated. Pseudopheochromocytoma is sometimes treatable by antihypertensive drugs or psychotherapy.
Pathology
Sporadic pheochromocytomas are generally solitary, well-circumscribed, encapsulated tumors (Fig. 109-5A).80 They usually are located in the adrenal gland or in its immediate vicinity. However, the adrenal gland may not be in its expected place atop the kidney, but actually may be located anywhere superior, inferior, medial, lateral, dorsal, or ventral to the kidney. Thus, pheochromocytoma may still be considered intra-adrenal in origin if the cortex of the adrenal is found in close relationship to the pheochromocytoma. Malignant tumors appear to be larger, to contain more necrotic tissue, and to be composed of smaller cells than are benign adrenal pheochromocytomas.18,81 However, it is difficult, if not impossible, to distinguish malignant from benign pheochromocytomas on the basis of histopathologic features. Capsular invasion, vascular penetration, coarse nodularity, the presence of atypical nuclei, higher mitotic count, absence of intracytoplasmic hyaline granules, and mitosis exist in both types of pheochromocytoma.80 Only tumor invasion of tissues and the presence of metastatic lesions (most commonly in the liver, lungs, lymphatic nodes, and bones) are consistent with the diagnosis of malignant pheochromocytoma.18,82 As described by Linnoila and colleagues, fewer neuropeptides are expressed in malignant than in benign pheochromocytoma cells.83 No differences have been noted in immunohistochemical expression of cathepsins, basic fibroblastic growth factor, c-met, or collagenase between benign and malignant pheochromocytomas.84 Clarke and colleagues recently reported that MIB-1 appears to be a good indicator of the potential of metastatic pheochromocytoma, with a specificity of 100% and a sensitivity of 50%.84
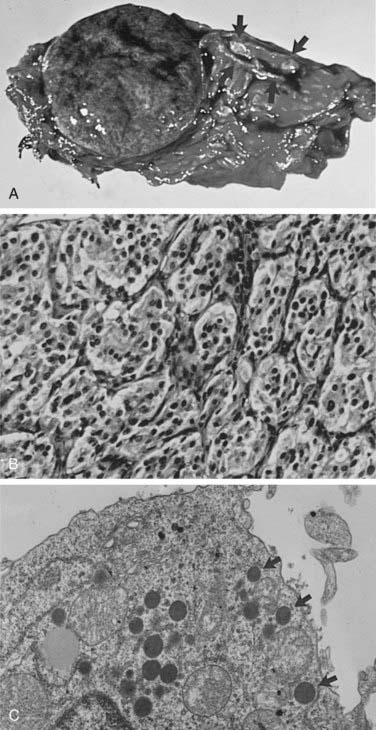
FIGURE 109-5. A, Cross section of a well-demarcated intra-adrenal pheochromocytoma with stippled areas of congestion. The normal adrenal cortex, which was a characteristic yellow-orange when fresh, is marked with arrows. B, The typical histology of an adrenal pheochromocytoma is shown with cells in nests and trabeculae separated by a thin vascular stroma (hematoxylin-eosin, ×400). C, Electron microscopy view of an adrenal pheochromocytoma with many large core catecholamine-containing granules, some marked with arrows. These granules, with a thin uniform halo between the core and the investing membrane, are the epinephrine-containing type. Granules with a prominent eccentric lucent space between the core and the limiting membrane are the norepinephrine-containing type (×13,500).
Most pheochromocytomas range in size from 3 to 5 cm.18,52,80 The largest tumor reported was 20 cm in diameter.85 Tumors often show areas of hemorrhage, as well as areas of necrosis with cystic degeneration. These areas of hemorrhage and necrosis are the cause of the inhomogeneity seen in the images of pheochromocytoma on computed tomography (CT) scans.
The chromaffin reaction, originally described by Henle in 1865, is a deep brown color of the adrenal medulla that occurs at least 12 hours after the tissue is placed in a dichromate solution.86 The reaction is caused by the oxidation of the catecholamines epinephrine and norepinephrine into adrenochrome pigments. When this pattern is well developed, it mimics tumor cell nests or “zellballen,” seen also in parasympathetic paragangliomas in the head and neck. Another pattern consists of anastomosing cords of cells (trabecular). The third and most common pattern is a mixture of anastomosing cell cords and nests of cells (see Fig. 109-5B).80 The tumor cells are usually polygonal with an intermediate amount of lightly colored eosinophilic granular cytoplasm. Cells may vary in size from small to large. Nuclei are well demarcated and generally eccentric in location. Nuclear pleomorphism with enlargement and hyperchromatism may be seen80; this is not diagnostic of malignancy. Occasionally, tumor cells resemble ganglion cells with rounded, eccentric nuclei and prominent nucleoli.
Touch preparations of the freshly cut surface of the tumor can be dried for several days over sodium hydroxide or another drying agent and then exposed to formaldehyde vapor. Examination under ultraviolet light reveals brightly colored catecholamine-containing granules within the cells of a pheochromocytoma.
Electron microscopy reveals the presence of membrane-bound, dense-core, neurosecretory granules 150 to 250 nm in diameter (see Fig. 109-5C).80 In most tumors, the predominant granule is the one associated with norepinephrine, whereas in the normal gland, the predominant granule is the one associated with epinephrine.87
Chromaffin cells have the ability to synthesize and secrete various amines and certain peptide hormones (i.e., ACTH, chromogranins, neuropeptide Y, calcitonin, angiotensin-converting enzyme, renin, vasoactive intestinal polypeptide, adrenomedullin, enkephalins, and atrial natriuretic factor), and this accounts for some of the different presentations of pheochromocytoma (e.g., Cushing’s syndrome, watery diarrhea). Antigens,88 protein gene product 9.5,89 galanin,90 renin,91 angiotensin-converting enzyme,92 and synaptophysin93 have been found in neuroendocrine neoplasms, including pheochromocytoma.
Genetics of Pheochromocytoma
Up to 24% of pheochromocytomas are inherited.12,18,46,52,61 Hereditary pheochromocytoma is associated with multiple endocrine neoplasia type 2 (MEN2A or MEN2B), von Recklinghausen’s neurofibromatosis type 1 (NF-1), von Hippel-Lindau (VHL) syndrome, and familial paraganglioma caused by germline mutations of genes encoding succinate dehydrogenase subunits B, C, and D (Table 109-4). In general, the traits are inherited in an autosomal dominant pattern.
Table 109-4. Pathologic Conditions Associated With Pheochromocytoma
| Syndrome | Genetic Abnormality | Phenotypic Abnormalities |
|---|---|---|
| Multiple Endocrine Neoplasia Syndromes | ||
| Multiple endocrine neoplasia type 2A (Sipple’s syndrome) | Chromosome 10 (10q11.2) | Medullary carcinoma of the thyroid |
| RET proto-oncogene mutations affect tyrosine kinase ligand-binding domain | Hyperparathyroidism | |
| Multiple endocrine neoplasia type 2B | Chromosome 10 (10q11.2) | Medullary carcinoma of the thyroid |
| RET proto-oncogene mutations affect tyrosine kinase catalytic site | Mucosal neuromas | |
| Intestinal ganglioneuroma | ||
| Megacolon | ||
| Marfanoid habitus | ||
| Neuroectodermal Syndromes | ||
| Neurofibromatosis (von Recklinghausen’s disease) type 1 (NF-1) | Chromosome 17 (17q11) mutations affect NF-1, tumor suppressor gene | Peripheral neurofibromas |
| Cerebelloretinal hemangioblastomatosis (von Hippel-Lindau syndrome, VHL) type 2 | Chromosome 3 (3p25-26) missense mutations affect VHL, tumor suppressor gene | Retinal angiomas |
| Cerebellar and spinal cord hemangioblastomas | ||
| Renal cell cancer | ||
| Pancreatic, renal, epididymal, and endolymphatic cysts/tumors | ||
| Succinate Dehydrogenase Gene Family Syndromes | ||
| SDHB: paraganglioma (PGL) type 4 | Chromosome 1 (1p36) missense, nonsense, and frameshift mutations | (Maligant) pheochromocytoma, parasympathetic paraganglioma |
| SDHC: PGL type 3 | Chromosome 1 (1q21) | Parasympathetic paraganglioma, pheochromocytoma (rare) |
| SDHD: PGL type 1 | Chromosome 11 (11q23) missense, nonsense, and frameshift mutations maternal imprinting | Parasympathetic paraganglioma, pheochromocytoma |
MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
MEN2 is an autosomal dominantly inherited syndrome (Sipple’s syndrome) that consists of pheochromocytoma, medullary carcinoma of the thyroid, and hyperparathyroidism.94–96 It affects about 1 in 40,000 individuals and is characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia/adenoma.97
The gene responsible for MEN2 is a proto-oncogene called RET.98 In contrast to MEN1, RET is specifically expressed in neural crest–derived cells such as calcitonin-producing C cells in the thyroid gland and catecholamine-producing chromaffin cells in the adrenal gland. Whether it is also expressed in the parathyroid glands remains to be ascertained, especially when the low rate of hyperparathyroidism in patients with MEN2A and the lack of hyperparathyroidism in those with MEN2B are considered, although both conditions are caused by mutations in the RET gene. RET plays a role in normal gastrointestinal neuronal and kidney development, as exemplified by the RET knockout mouse, which has a Hirschsprung-like phenotype and renal cyst or agenesis.99 RET is located on chromosome 10q11.2 and encodes a receptor tyrosine kinase, RET protein. As an oncogene, activation of RET leads to hyperplasia of target cells in vivo. Subsequent secondary events then lead to tumor formation.100 RET consists of 21 exons with 6 so-called “hot spot exons” (exons 10, 11, 13, 14, 15, and 16) in which mutations are identified in 97% of patients with MEN2. Recently, it has been proposed that in addition to other events, RET protein accumulation, secondary to absent or reduced VHL protein, may then cause transformation of selected chromaffin cells to pheochromocytoma.100 Additional studies are needed to clarify whether such somatic VHL gene alterations in MEN2-associated tumors play a role in early or late tumorigenesis or rather in tumor progression. RET germline mutation screening is commercially available (in the US, http://endocrine.mdacc.tmc.edu; Mayo Clinic, Rochester, MN) and has widely replaced the cumbersome provocative testing of calcitonin stimulation (with calcium and/or pentagastrin), which has been unreliable.
Pheochromocytomas (at least 70% of which are bilateral) develop on a background of adrenomedullary hyperplasia and become manifest (e.g., biochemically or on imaging) in about 50% of patients. MEN2-associated pheochromocytomas are almost exclusively benign (with <5% reported to be malignant) and localized to the adrenals. The peak age is around 40 years, but children as young as 10 years can be affected.101,102
Patients with MEN2-related pheochromocytoma often lack sustained hypertension or other symptoms (they occur only in about 50%). Because MEN-related pheochromocytomas secrete epinephrine, stimulation of β-adrenergic receptors causes palpitations and tachycardia. Therefore, their detection is based mainly on elevated plasma metanephrine and epinephrine levels. In patients with pheochromocytomas that exclusively produce normetanephrine, MEN2 can be excluded. MEN2-related pheochromocytomas are almost always intra-adrenal, are often bilateral (≈30% at diagnosis), and are rarely malignant (<5%).46,103 In addition, as with most epinephrine-secreting pheochromocytomas, the hypertension is more likely to be paroxysmal than sustained. For these reasons, the diagnosis is easy to miss.
Medullary thyroid carcinoma is found in most patients with MEN2A, pheochromocytoma in a somewhat lesser percentage, and hyperparathyroidism in only about 25% of affected patients. Pheochromocytomas in MEN 2A most often are diagnosed at between 30 and 40 years of age. The syndrome results from germline mutations in the RET proto-oncogene on chromosome 10 (10q11.2).98
Patients with MEN2B have pheochromocytoma, medullary carcinoma of the thyroid, ganglioneuromatosis, multiple mucosal neuromas of eyelids, lips, and tongue, and some connective tissue disorders that include marfanoid habitus, scoliosis, kyphosis, pectus excavatum, slipped femoral epiphysis, and pes cavus.104 This syndrome also appears to be caused by germline mutations in the RET proto-oncogene on chromosome 10, but these mutations affect the tyrosine kinase catalytic site of the protein.105 In children with MEN2B-associated pheochromocytomas, a higher risk of malignancy compared with MEN2A or sporadic disease is found.106
MEN1 (Wermer’s syndrome) consists of hyperparathyroidism, pituitary adenomas, and pancreatic islet cell tumors. Pheochromocytoma is not usually part of this complex; however, the occurrence of pheochromocytoma and pancreatic islet cell tumors has been reported in some families.107 Often, the islet cell tumors are nonfunctional. Various “crossover” syndromes have been reported in which pheochromocytoma has been associated with characteristics of MEN1, MEN2A, MEN2B, von Recklinghausen’s neurofibromatosis (NF), VHL, and the Zollinger-Ellison syndrome.108 It has been proposed that the combination of neurofibromatosis, duodenal carcinoid, and pheochromocytoma constitutes a neuroendocrine syndrome that is separate from the combination of VHL, islet cell tumor, and pheochromocytoma.109
VON HIPPEL-LINDAU SYNDROME
Another neuroectodermal syndrome commonly associated with pheochromocytoma is VHL syndrome, which is caused by mutations in chromosome 3 (3p25-26), which encodes the VHL tumor suppressor gene.110 Pheochromocytomas in VHL disease typically develop according to Knudson’s two-hit model, including an inherited germline mutation of VHL and loss of function of the wild-type allele of the VHL gene. The disease has been divided into two types based on the significant genotype-phenotype correlations observed. Type 1 includes mainly large deletions or mutations and expresses the full phenotype of vascular lesions of the retina, cysts or solid tumors in the brain or spinal cord, pancreatic cysts, renal cell carcinoma, epididymal cystadenoma, and endolymphatic sac tumors, but no pheochromocytoma. Type 2 has missense mutations, pheochromocytoma, and the full phenotype.111 Patients often are asymptomatic when they present with other aspects of this disease. This syndrome is variable in terms of the different organ systems involved and the extent of involvement from patient to patient and from family to family. Overall, less than 30% of patients with a VHL germline mutation develop a pheochromocytoma. Pheochromocytomas as part of the VHL syndrome have an exclusively noradrenergic phenotype, reflecting the production of norepinephrine only.112 These tumors are located mainly intra-adrenally and are bilateral in about 50% of patients, with a less than 7% incidence of metastases. Because they do not express glucagon receptors, the glucagon test should not be used for detection of these tumors. These tumors are commonly found on the basis of periodic annual screening, or during searches for other tumors that are part of this syndrome. Therefore, when detected, these tumors are commonly small; they often fail to be detected by nuclear imaging methods. Furthermore, about 80% of pheochromocytomas found in patients with VHL during screening are asymptomatic and are not associated with hypertension.
NEUROFIBROMATOSIS TYPE 1
Von Recklinghausen’s NF is now divided into two types: NF-1 has neurofibromas of peripheral nerves, whereas NF-2 has central neurofibromas. NF-1 is inherited in an autosomal dominant pattern. The association with pheochromocytoma is one between a relatively common disease and a rare disease. Thus, although less than 1% to 2% of patients with NF have pheochromocytoma, about 5% of patients with pheochromocytoma have NF.113 Pheochromocytoma associated with NF-1 is caused by germline mutations in chromosome 17 (17q11), where the NF-1 gene encodes for the protein neurofibromin. These mutations lead to inactivation of this tumor suppressor gene and its protein.114 Similar mutations introduced into the NF-1 gene in mice lead to pheochromocytoma, which is otherwise rare in these animals.115 Pheochromocytoma in patients with NF-1 is rarely seen in children, because it usually occurs at a later age (around 50 years). Only 12% of patients with NF-1 are diagnosed with bilateral and multifocal pheochromocytomas, and less than 6% of patients have metastatic pheochromocytoma.116
The incidence of pheochromocytoma in NF-1 is relatively low (about 1%) compared with other hereditary syndromes, and routine screening of such patients generally is not recommended. However, if a patient with NF-1 has hypertension, then a pheochromocytoma should be considered and excluded.113
SUCCINATE DEHYDROGENASE GENE FAMILY
Recently, pheochromocytoma susceptibility has been associated with germline mutations of the succinate dehydrogenase (SDH) gene family.117,118 The SDH genes (SDHA, SDHB, SDHC, and SDHD) encode the four subunits of complex II of the mitochondrial electron transport chain,119 which is essential for the generation of ATP (oxidative phosphorylation). SDHB and SDHD mutations can lead to complete loss of SDH enzymatic activity, a phenomenon that has been linked to tumorigenesis through upregulation of hypoxic-angiogenetic responsive genes.120 Unlike the SDHA gene, mutations of SDHB, SDHC, and SDHD genes are associated with the presence of familial and nonfamilial pheochromocytoma and parasympathetic paraganglioma. Frameshift, missense, and nonsense mutations were identified for the SDH gene family. In recent studies, it has been found that about 4% to 12% of sporadic pheochromocytomas12,121 and up to 50% of familial pheochromocytomas121 have SDHD or SDHB mutations. The SDHB, SDHC, and SDHD traits are inherited in an autosomal dominant fashion and give rise to familial paraganglioma (PGL) syndromes 4, 3, and 1, respectively (see Table 109-4). The penetrance of these traits is incomplete, however. Furthermore, SDHD-related disease is characterized by maternal genomic imprinting. Because of silencing of the maternal allele by methylation, individuals who inherit a mutation from their mother remain free of paraganglioma but still may pass on the mutation to their offspring. SDHB mutations predispose to mainly extra-adrenal pheochromocytomas with a high malignant potential, and less frequently to benign parasympathetic head and neck paragangliomas.62,122–124 SDHD mutations typically are associated with multifocal parasympathetic head and neck paragangliomas and usually with benign extra-adrenal and adrenal pheochromocytomas.122,124 Metastastic pheochromocytoma is rare in SDHD mutation carriers, but it can occur.123,125 SDHC mutations are rare and are almost exclusively associated with parasympathetic head and neck paraganglioma,126 although rare cases of SDHC-associated extra-adrenal pheochromocytoma have been reported.127,128 Most SDHB-related paragangliomas secrete norepinephrine or both norepinephrine and dopamine,62 a profile that is consistent with mainly extra-adrenal tumors. Some SDHB-related PGLs exclusively overproduce dopamine, but not other catecholamines 62,129–131 Therefore, measurement of plasma levels of dopamine or its O-methylated metabolite methoxytyramine should be considered in SDHB-related pheochromocytoma. As mentioned, ≈10% of SDHB-related sympathetic paragangliomas are “biochemically silent.” The biochemical phenotype of SDHD-associated pheochromocytoma has not been studied consistently.
We recommend to offer genetic testing, preceded by careful genetic counseling, to all first-degree family members of patients with SDH-related pheochromocytoma and paraganglioma,132 and to subsequently carry out a periodic clinical, biochemical, and radiologic tumor screening among mutation carriers, especially those with an SDHB mutation. No evidence-based protocol has been used for tumor screening among children with a known SDHB, or any other mutation. We recommend periodic biochemical and total body magnetic resonance imaging (MRI) screening from as early as age 7.
SPORADIC AND OTHER PHEOCHROMOCYTOMAS
Genetic analyses of DNA from sporadic pheochromocytoma have yielded variable results, with up to 20% RET mutations (half MEN2A and half MEN2B) in some series, and up to 20% VHL mutations in others.46,133,134 In another study, 45% of sporadic adrenal pheochromocytomas had loss of heterozygosity at the VHL gene locus, but no genetic abnormalities were found in sporadic extra-adrenal pheochromocytomas.135 Genetic analysis provides positive identification of carriers in familial syndromes and thus identifies who should be screened and who need not be screened for the various traits of the phenotype.
Pheochromocytoma also may occur as part of Carney’s triad (i.e., gastric leiomyosarcoma, pulmonary chondroma, and extra-adrenal pheochromocytoma).136 The syndrome is very rare; fewer than 30 cases have been reported, and only 25% of patients manifest all three parts of the triad. It occurs sporadically and is clearly nonfamilial.137 Recently, a new syndrome called the Stratakis-Carney syndrome, or the “dyad of gastrointestinal stromal tumor and paraganglioma” associated with SDHB, SDHC, and SDHD mutations, was identified.138
For the genetic diagnosis of pheochromocytoma, a panel of experts at the International Symposium on Pheochromocytoma 2005 held in Bethesda, Maryland, agreed that, although there is now a reasonable argument for more widespread genetic testing, it is neither appropriate nor currently cost-effective to test for every disease-causing gene in every patient with a pheochromocytoma or paraganglioma. Rather, it was stressed that the decision to test, and which genes to test, requires judicious consideration of numerous factors, several of which are noted in Fig. 109-6.
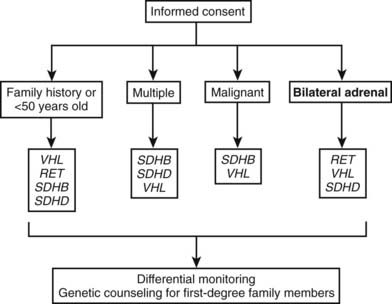
FIGURE 109-6. Algorithm for genetic testing for genes associated with pheochromocytoma. The algorithm should be applied if there is a family history of pheochromocytoma, the patient is <50 years old, or multiple malignant or bilateral tumors are present. The biochemical phenotype of a tumor should also be considered in selection of the most appropriate genes to test. The term multiple in the figure indicates tumors in separate anatomic locations. Patients with multiple endocrine neoplasia type 2 might have multiple tumors in a single adrenal gland. It should be considered that multiple or bilateral tumors often do not occur simultaneously. RET, Rearranged during transfection; SDHB, succinate dehydrogenase subunit B gene; SDHD, succinate dehydrogenase subunit D gene; VHL, von Hippel-Lindau gene.
Biochemical Diagnosis of Pheochromocytoma
INITIAL BIOCHEMICAL TESTING
Missing a pheochromocytoma can have deadly consequences. Therefore, one of the most important considerations in the choice of an initial biochemical test is a high level of reliability that the test will provide a positive result in that rare patient with the tumor. Such a test also provides confidence that a negative result reliably excludes the tumor, thereby avoiding the need for multiple or repeat biochemical tests or even costly and unnecessary imaging studies to rule out the tumor. Therefore, the initial workup of a patient with suspected pheochromocytoma should include a suitably sensitive biochemical test.
Catecholamine secretion by pheochromocytomas can be episodic or, in patients who are asymptomatic, negligible in nature. Thus, measurements of urinary or plasma catecholamines do not provide reliable biochemical tests for detection of the tumor. This is particularly problematic during screening for pheochromocytoma in patients with a predisposing hereditary condition, where 26% to 29% of tumors can be expected to be associated with normal urinary excretion or plasma concentrations of catecholamines.139 Because of the intermittent nature of catecholamine secretion, it has been recommended that urine or blood samples are best collected when the patient is hypertensive. Hypertension, however, can be present in some patients independently of catecholamine release by a tumor. Thus, normal catecholamines in a patient with hypertension do not exclude pheochromocytoma.
Because metanephrines are produced continuously within pheochromocytoma tumor cells, and independently of catecholamine release, these measurements obviate any need for collection of blood or urine samples during hypertensive episodes. This understanding has provided a rationale for subsequent studies examining the utility of measurements of plasma free metanephrines for diagnosis of pheochromocytoma.8,9,139–144 At the National Institutes of Health (NIH), the superiority of plasma free metanephrines over other tests was established in a series of three published reports,9,139,143 culminating in a study involving more than 850 patients, including 214 with pheochromocytoma.9 Plasma free metanephrines showed superior combined diagnostic sensitivity and specificity over all other tests examined, including urinary and plasma catecholamines, urinary vanillylmandelic acid, and urinary total and fractionated metanephrines. Analysis of receiver-operating characteristic curves showed that at equivalent levels of sensitivity, the specificity of plasma free metanephrines was higher than that of all other tests, and that, at equivalent levels of specificity, the sensitivity of plasma free metanephrines was also higher than that of all other tests, even when the latter were combined.
The high diagnostic sensitivity of measurements of plasma free metanephrines has now been independently confirmed by four other groups of investigators.8,140,142 All together, the five independent studies—with 336 patients with pheochromocytoma and close to 2500 patients in whom the tumor was excluded—have indicated an overall diagnostic sensitivity of 98% and specificity of 92% for measurement of plasma free metanephrines.
Metanephrines in urine usually are measured after a deconjugation step that liberates free metanephrines from sulfate- or glucuronide-conjugated metanephrines,145,146 whereas plasma metanephrines are measured most often in the free form, but they can also be measured after a deconjugation step (Fig. 109-7). Conjugated metanephrines are present in urine and plasma at much higher concentrations than are free metanephrines. Adding to the confusion in terminology is the fact that historically metanephrines were measured in urine by spectrophotometric methods as “total” metanephrines, representing the combined sum of deconjugated normetanephrine (the O-methylated metabolite of norepinephrine) and deconjugated metanephrine (the O-methylated metabolite of epinephrine).
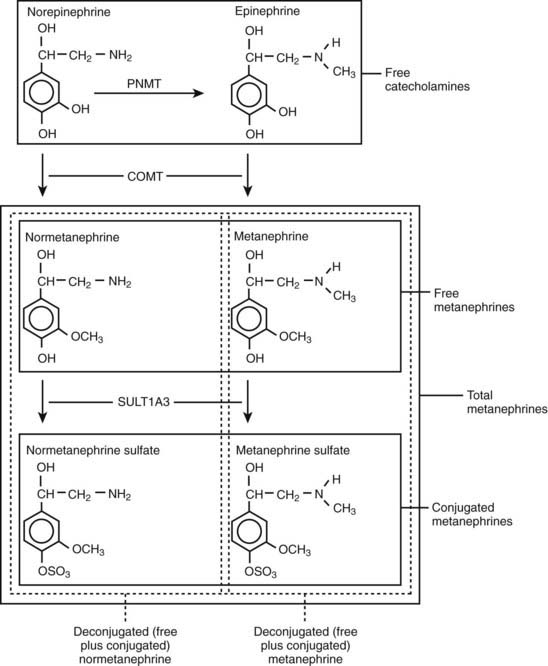
FIGURE 109-7. Pathway of production of free and sulfate-conjugated metanephrines, illustrating differences in free metanephrines measured in plasma and deconjugated metanephrines (free plus conjugated metanephrines) measured in urine. Deconjugated metanephrines in urine may be measured as fractionated metanephrines (i.e., separate measurements of deconjugated normetanephrine and metanephrine) or as total metanephrines (i.e., the combined sum of deconjugated normetanephrine and metanephrine). COMT, Catechol O-methyltransferase; PNMT, phenylethanolamine N-methyltransferase; SULT1A3, monoamine-preferring phenol sulfotransferase.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


