FIGURE 71-1. Radiograph demonstrating osteoporosis circumscripta of the skull affecting the frontal and temporal regions.

FIGURE 71-2. Radiograph of an osteolytic lesion of Paget’s disease that began in the diaphysis of the ulna and exhibits the characteristic flame-shaped or inverted-V extension toward the wrist. Note also the expansile nature of the lesion.
A more commonly observed stage of Paget’s disease is the mixed phase, in which osteoblastic (or osteosclerotic) features are intermixed with osteolytic features in an individual bone. This phase is best appreciated in long bones, in which one may observe the advancing osteolytic front adjacent to normal bone and, trailing this front, a heterogeneous region of osteosclerosis superimposed on the region that had previously been dominated by the osteolytic process (Fig. 71-3). Biopsies of the mixed phase reveal a characteristic abnormality of lamellar bone in both cortical and trabecular bone. The matrix is transformed into a bizarre “mosaic” pattern of irregularly juxtaposed pieces of lamellar bone separated by cement lines that have a scalloped outline. The irregularity probably reflects areas of previous osteoclastic resorption. The structure of the involved cortex is so disordered that complete osteons are rare, and the outer and inner circumferential lamellae and the interstitial lamella may be totally disrupted.
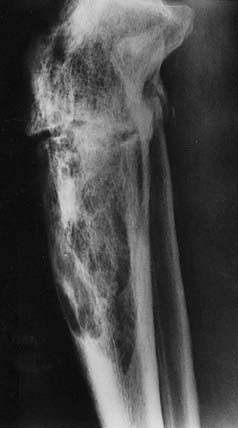
FIGURE 71-3. Radiograph of a tibia exhibiting a distal advancing osteolytic front with proximal sclerotic bone. A partially healed pathologic fracture is present proximally.
The same disordered matrix structure is seen in trabecular bone. Interspersed among the chaotic lamellae are patches of woven bone characterized by a random pattern of deposition of collagen fibers and a larger number of osteocytes per unit area of matrix. It has been suggested that the lacunae surrounding the osteocytes are larger than normal and that this finding represents osteocytic osteolysis.33 However, it is more likely that the increased size of the periosteocytic lacunae is simply a characteristic of woven bone and not a second type of bone resorption in Paget’s disease. At the surfaces of bone formation, plump osteoblasts are found in great number adjacent to abundant osteoid. This type of bone is seldom found in adults except when associated with rapid remodeling of bone, such as occurs after a fracture or in response to tumor invasion of bone. Studies using quantitative histomorphometry of bone documented the marked degree of cellular activity underlying the dramatic changes in bone structure in Paget’s disease.34 The total amount of osteoid and the percentage of the bone surface covered by osteoid may be increased fourfold to fivefold. The increase in osteoid is not associated with an increase in osteoid seam width because the rate of calcification also is increased, as established by double labeling of bone-forming surfaces with tetracycline. No dynamic means of defining the rate of bone resorption is available, but the extent of the total bone surface exhibiting evidence of bone resorption averages about sixfold that of normal individuals, and the number of osteoclasts may be increased as much as 10-fold. In the medullary cavities, the intense resorptive process may produce hemorrhagic cysts with encircling fibrous marrow containing macrophages filled with hemosiderin. These cysts are believed to result from rupture of multiple dilated vessels and ensuing microinfarctions.35 In the mixed phase of Paget’s disease, not only does patchy sclerosis of bone become apparent on radiography, but a bone also may be enlarged. If the osteolytic process extends to the subperiosteal layer, bone formation may be stimulated to such an extent that the thickness and circumference of the bone are increased as a result of periosteal new bone formation. When the skull is affected, this process can produce as much as a fourfold thickening of the calvarium, as was reported by Paget.36 A patchy form of sclerosis is often of a “cotton-wool” character (Fig. 71-4). The skull may be so severely affected that platybasia, or basilar impression, may be a complication. Long bones may be shortened, and typically lateral bowing of the femur or anterior bowing of the tibia or both may develop. Later in the course of the disease, the tibia may exhibit severe lateral bowing. The pathogenesis of the slowly progressive deformity is not known but must be related to the state of abnormal remodeling of the bone. Frequently, fissure fractures are associated with the bowing deformity. These fractures are linear transverse radiolucencies that usually are present in the cortex of the convex aspect of the deformed bone. They often are multiple and may remain stable in appearance for years. They can be found in either osteopenic or osteosclerotic cortices and may be present even in the absence of deformity. Histologic examination of these lesions suggests that they are incomplete fractures.2 Only a small percentage of these lesions progress to a complete transverse fracture, which has been seen more often in patients with a sclerotic cortex.
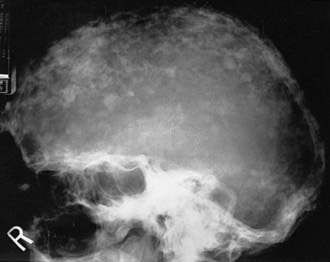
FIGURE 71-4. Radiograph of the sclerotic phase of Paget’s disease in the skull exhibiting a “cotton-wool” appearance.
Even after osteosclerotic bone has invaded the previously osteolytic regions of affected bone, evidence of ongoing abnormal bone resorption can be seen in the form of secondary resorption fronts. These secondary fronts are commonly seen as clefts in the cortex of long bones and trail in the path of the primary front.
In the final stage of Paget’s disease, termed osteoblastic or sclerotic, the affected bone remains dense and retains the “mosaic” matrix pattern characteristic of Paget’s disease. Tubular bones show a loss of differentiation between cortical and trabecular bone, even with enlargement of the bone as a result of periosteal new bone formation, because the new bone is no longer compact bone. Much less cellular activity is present in sclerotic lesions. Osteoclasts are few or absent, but osteoblasts may still be seen to line bone surfaces. The marrow may remain fibrous, but the numbers of blood vessels are greatly reduced. Scattered chronic inflammatory cells may be present. Occasionally, parts of lesions are totally devoid of bone cells, and for this reason, the concept of “burned-out” Paget’s disease has arisen. However, it is very unlikely that extensive lesions of Paget’s disease ever achieve an entirely burned-out state. On the contrary, the presence of all stages of the disease in a single bone is much more likely.
The evolution of Paget’s disease can also be observed by administration of radioactive tracers and scanning of the entire skeleton or selected regions. Bone scans use technetium-labeled bisphosphonates, which after intravenous injection localize to skeletal sites in proportion to the relative blood flow and the rate of bone formation. The scans usually, but not always, demonstrate high uptake of radioactivity in the areas of the skeleton noted to be radiographically abnormal37 (Fig. 71-5). In a small proportion of patients, increased uptake may be seen when the radiograph is normal, thus illustrating the great sensitivity of bone scans. On the contrary, a small percentage of sclerotic lesions may not be picked up by a bone scan. These lesions appear to represent areas of inactive disease. Bone scans can be analyzed semiquantitatively or by computer and thus may be used to monitor response to treatment.
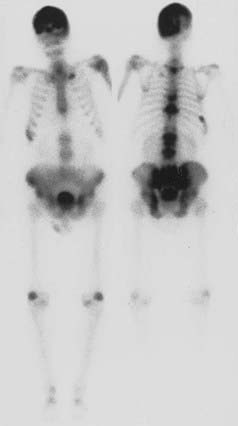
FIGURE 71-5. Anterior and posterior views of a bone scan in a patient with polyostotic Paget’s disease. Increased uptake of tracer can be noted in the skull, multiple vertebrae, and the pelvis, areas in which Paget’s disease was observed on radiographs. The other abnormal areas probably represent degenerative arthritis and a healed rib fracture.
Gallium scans, most often used to detect occult infection or tumors, also have been shown to delineate the lesions of Paget’s disease.38 Evidence indicates that tracer gallium is localized to the nuclei of osteoclasts.39 Therefore, the gallium scan may serve as a direct index of cellular activity in Paget’s disease.
Clinical Features
A considerable proportion of individuals with Paget’s disease have neither symptoms nor signs of the disease.1 The disease is accidentally discovered in these cases because of radiologic or biochemical abnormalities uncovered during investigation of another disorder.
The most common clinical problems are pain and deformity. The bone pain of Paget’s disease, when present, is seldom severe. Usually it is a dull pain, is located deep below the soft tissues, and often persists during the night. In weight-bearing bone, the pain may be slightly worsened by ambulation, but to a lesser extent than pain originating from the joints or from nerve impingement. Deformity of the skeleton is most often noted in the skull and the lower extremity. Over many years, the hat size of an affected individual may be noted to increase. Bowing and enlargement of the femur and tibia also may evolve over a period of years.
REGIONAL MANIFESTATIONS
The Skull
In the absence of an enlarged cranium, symptoms in the skull are uncommon. Even with an enlarged cranium, symptoms often are absent. Certainly, the most common symptom (30% to 50%) is hearing loss,40 which is slowly progressive in untreated patients. Vertigo or tinnitus or both are much less common. The main mechanism of hearing loss in Paget’s disease has been attributed to a reduction in bone mineral density of the cochlear capsule.41
Much more serious complications of Paget’s disease may occur in the advanced stage of the disease in a small number of patients. The weight of the skull may be so great that the ability of the patient to keep the head erect is impaired. Muscle spasm may then produce pain in the neck and tension headaches. Neurologic abnormalities may be found in such patients as a consequence of basilar invagination or platybasia. Although they are unusual complications, even in the presence of radiologic evidence of basilar invagination, compression of structures in the posterior fossa or cerebellar tonsillar herniation may produce ataxia, muscle weakness, and impaired respiration. Hydrocephalus also has been noted to be a rare complication and may be manifested by impaired gait, urinary incontinence, and some degree of dementia.
Finally, severe skull disease may be associated with the vague findings of a withdrawn individual who is somnolent and weak. It has been suggested that this manifestation might be a consequence of shunting of blood from the brain vessels to the external carotid artery system, a possible pagetic steal syndrome.42 These symptoms also could represent a psychological response to disability, inasmuch as nearly 50% of patients in one study have been reported to have depression.43
The Jaws
Paget’s disease may affect the facial bones and jaws, but such involvement is uncommon. Leontiasis ossea is the descriptive term applied to a patient with enlargement of all the facial bones, but such deformity is more likely to be found in fibrous dysplasia.
Involvement of the mandible or maxilla may produce progressive root resorption leading to the loss of teeth.44 In the more advanced stages of Paget’s disease, excessive formation of the cementum is associated with absence of the lamina dura and periodontal membranes. Facial disfigurement may occur from enlargement of the maxilla or mandible or both and is associated with spreading of the teeth and malocclusion. Edentulous patients have difficulty acquiring properly fitting dentures. Oral surgery may be complicated by excessive intraoperative bleeding and postoperative osteomyelitis. Tooth extractions may prove difficult because of ankylosis resulting from hyperplasia of the cementum.
The Spine
Neck pain and back pain are common complaints in an aging population of patients with Paget’s disease. Visualization of Paget’s disease in one or more vertebrae on radiographic examination often leads clinicians to conclude that they are dealing with bone pain, and treatment is instituted. However, most patients with moderate to severe pain have a complication associated with Paget’s disease as the cause of the pain rather than bone pain alone.
Paget’s disease affects the lumbar and sacral regions most frequently. One vertebra or many vertebrae may be involved. In the early osteolytic phase, which often is not recognized, the vertebral body appears osteoporotic and, in rare cases, may undergo so much resorption that it takes the shape of a thin transverse rod. Much more often, the vertebral bodies become enlarged overall, with thickened margins and coarse vertical striations centrally. Compression of a sclerotic vertebral body may develop because of the abnormal mechanical properties produced by the chaotic microarchitecture.
Severe pain or impaired neurologic function or both may result from compression of the spinal cord or nerve roots.45 This complication can arise from enlargement of the vertebral bodies, pedicles, or laminae, as well as from compression fractures. It also has been suggested that shunting of blood may occur from the spinal arteries to the highly vascular bone.46 Neurologic syndromes are more likely to develop with thoracic involvement. Symptoms include back pain, difficult ambulation, numbness, paresthesias of the feet, and progressive paresis of the legs. Later problems can include impaired bladder and bowel function, as well as spastic paraparesis and loss of sensation. Computed tomography (CT) and magnetic resonance imaging (MRI) are particularly helpful in resolving the anatomic abnormalities producing the disturbed function.
A rare complication in the spine is the development of a discrete paraspinal mass consisting of a central marrow cavity surrounded by pagetic bone that extends from the vertebrae.47 It may appear that the lesion represents a neoplasm, but careful analysis of prior radiologic studies may reveal the chronic nature of the lesion and may make it unnecessary to perform a biopsy.
The other major causes of back pain in Paget’s disease are intervertebral disk disease and degenerative arthritis. No evidence indicates that disk degeneration is more common in patients with Paget’s disease, but it has been reported that the pagetic process can invade the disk and produce bony bridging across the disk space.48 Back pain in the lumbar region is frequently associated with degenerative arthritis,49 particularly when distortion of the facet joints is associated with Paget’s disease. Large osteophytes also may be found in association with enlarged vertebral bodies or where a compression fracture has occurred. A syndrome mimicking ankylosing spondylitis may occur in the presence of extensive osteophyte formation or with ossification of spinal ligaments,50 but the human leukocyte antigen (HLA)-B27 antigen is absent. However, classic ankylosing spondylitis has been found in association with Paget’s disease.49
The Pelvis and Extremities
The main symptoms associated with pelvic and lower extremity involvement are pain and impaired ambulation. Pain is seldom a significant symptom in the osteolytic phase of the disease. Hip pain is most common when both the acetabulum and the proximal end of the femur are affected by the sclerotic phase of the disease.50 Bowing of the femur and protrusio acetabuli are often associated with pain aggravated by weight bearing. Many patients are relatively comfortable when not weight bearing, unlike patients with bone pain, who usually have nocturnal discomfort.
Knee pain and occasionally joint effusions may occur with sclerotic disease affecting the femur or tibia or both. Distortion of the knee joint produced by enlargement of the distal end of the femur or the proximal part of the tibia and severe bowing of either bone can induce mechanical strains on the articular cartilage and thereby accelerate the degenerative process. The pagetic process in subchondral bone also may contribute to joint disease. A similar set of circumstances may account for ankle pain.
Fractures of the lower extremity are more likely to affect the femur than the tibia. In the largest series of reported femoral fractures, the subtrochanteric region was the most common site of fracture (49 of 182), and the rate of nonunion was noted to be 40%, a figure considerably greater than was previously appreciated.51 Nonunion appears to be less common after tibial fractures.
Involvement of the upper extremity long bones is much less likely to produce symptoms, although deformity may be apparent. At the shoulder, impaired rotator cuff function may be noted when overgrowth of bone leads to anatomic distortion of the glenohumeral joint.
Occasionally, patients who have Paget’s disease affecting the foot have pain on weight bearing. Symptoms are seldom encountered in individuals with radiologic evidence of Paget’s disease in the hands.
SARCOMA, GIANT CELL TUMORS, AND NONSKELETAL MALIGNANCIES
The most feared complication in Paget’s disease is sarcoma. It has been estimated that 10% of patients with extensive disease may experience this problem,35 but if all affected individuals are considered, the incidence is probably less than 1%.52 Sarcomas have rarely been reported to develop in multiple members of families with Paget’s disease.
Patients in whom sarcomas develop usually have pain and swelling, always in an area previously affected with Paget’s disease. Occasionally, fracture at the tumor site may lead to discovery of the neoplasm. Tumors most often arise in the pelvis, femur, humerus, skull, and facial bones.53 Multifocal sarcomas are found only in patients with advanced and widespread polyostotic disease and are thought to represent tumors of independent origin rather than metastases.54
The histology of sarcomas is quite variable. Fibrosarcomas, chondrosarcomas, osteogenic sarcomas, and anaplastic sarcomas may be found.35 Variable numbers of multinucleated giant cells (probably osteoclasts) may be scattered throughout the tumor stroma and most likely are not neoplastic. The nuclear inclusions typical of Paget’s disease have been observed in giant cells but not in the tumor cells.55 It is not unusual for several histologic patterns to be present in a single tumor, which suggests that a common stem cell may give rise to a variety of better differentiated mesenchymal cells.
Lymphomas and multiple myelomas have been found in association with Paget’s disease52 but probably represent chance occurrence rather than a complication of the pathologic process.
It is difficult to detect early sarcoma formation by radiologic examination because of the underlying distortion of pagetic bone. Because they appear to arise in medullary bone, an early finding may be a subcortical osteolytic lesion. Only when a radiolucent focus with speckled areas of calcification has broken through the confines of the cortex is it apparent that a malignant neoplasm is present. CT or MRI is the best means of determining the extent of the tumor mass.
The rate of change in serum alkaline phosphatase activity has not proved to be a useful marker for the development of sarcomas despite early reports that this might be the case.
The life expectancy for the average patient in whom a sarcoma develops is sadly brief, perhaps because of the difficulty associated with early detection. In one study, only 7.5% of patients survived 5 years, whereas in elderly patients free of underlying Paget’s disease, a 37% 5 year survival rate was noted.56
Giant cell tumors of bone, which usually follow a benign course and most often are found at the ends of long bones in otherwise normal individuals, may arise in the lesions of Paget’s disease.57 They appear to be much less common than sarcoma in Paget’s disease and have frequently been noted to originate in the skull and facial bones. Rarely, these tumors have been reported in multiple family members who have Paget’s disease.
These tumors are characterized by spindle-shaped cells with fusiform nuclei and clumped chromatin or nuclei and by scattered multinucleated giant cells. Mitoses are rarely found in either the mononuclear or giant cells. The giant cells contain the nuclear inclusions of Paget’s disease, but the stromal cells do not.57 The opinion has been expressed that many of the reported cases of giant cell tumor in Paget’s disease actually represent giant cell reparative granulomas, which are common lesions arising in the jaw.58
Surgery and radiation have been used to treat symptomatic giant cell tumors in Paget’s disease, and in one patient, high doses of dexamethasone were effective in shrinking an extraskeletal tumor.59
Biochemical Features
The intense cellular activity in active lesions of Paget’s disease may be reflected in various biochemical markers of bone resorption and bone formation. In most patients who come to clinical attention, biochemical markers do reflect the extent and activity of the disease, although patients with only a small percentage of affected skeleton have no biochemical abnormalities.
INDICES OF BONE RESORPTION
The increased bone resorption typical of active Paget’s disease might be expected to produce an increase in serum and urinary calcium levels, but in the absence of fractures or immobilization, hypercalcemia or hypercalciuria is not a prominent feature of Paget’s disease.60 It is generally believed that this finding is explained by a concomitant increase in bone formation that is demonstrable histologically and by kinetic analysis of plasma disappearance rates and skeletal uptake of radiocalcium60 or other skeletal tracers. A variety of bone collagen matrix breakdown products have been used as indices of bone resorption. These products include urinary hydroxyproline, total and free pyridinoline and deoxypyridinoline, type 1 collagen N-telopeptide, and type 1 collagen C-telopeptide. The telopeptide assays appear to be most specific for bone collagen resorption. Urinary N-telopeptide excretion is sensitive to therapeutic intervention.61 Measurement of non-isomerized fragments of collagen type 1 C-telopeptide may be the most sensitive means of evaluating bone resorption in patients with Paget’s disease.62
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


