FIGURE 151-1. Distribution of C cells in the thyroid gland. A, Reconstruction of the distribution of C cells in the thyroid gland. The greatest concentration of C cells occurs at the junction of the upper third and lower two thirds of the gland. This distribution explains the characteristic location of hereditary medullary thyroid carcinoma. B, Hereditary medullary thyroid carcinoma is almost always bilateral, although the extent of involvement may not be equal. C, Sporadic medullary thyroid carcinoma is most commonly a unilateral process that may develop at any location within the thyroid gland.
(A, Adapted from Wolfe HJ, Voelkel EF, Tashjian AH Jr: Distribution of calcitonin containing cells in the normal adult human thyroid gland: a correlation of morphology with peptide content, J Clin Endocrinol Metab 38:688–694, 1974; B and C, From Grauer A, Raue F, Gagel RF: Changing concepts in the management of hereditary and sporadic medullary thyroid carcinoma, Endocrinol Metab Clin North Am 19:613–635, 1990.)
The adrenal chromaffin cell is thought to migrate from neural crest tissue,25 although this point has not been established as clearly as for the C cell. These cells uniquely express the enzyme norepinephrine N-methyltransferase (PNMT) and are capable of converting norepinephrine to epinephrine. Differentiation of the adrenal chromaffin cell is a complex process that is dependent on several signaling pathways, including transforming growth factor (TGF)-β2, TGF-β3, bone morphogenic protein (BMP)-4,26 and several members of the neurotropin receptor family.27,28 Earlier literature suggesting that glucocorticoids are important in differentiation of the adrenal medulla29 appears to be incorrect or at least incomplete.26 A transcription factor, MASH-1, is an important regulator of normal chromaffin cell differentiation. Mice deficient in this transcription factor have a reduced number of adrenal chromaffin cells, and those present have a reduced amount of tyrosine hydroxylase, which is necessary for catecholamine synthesis.30 Deletion of the RET receptor in mice has little effect on development of the adrenal medulla,31,32 although accumulating evidence suggests that expression of this receptor is necessary for PNMT expression, epinephrine production, and development of a neuronal phenotype.31,33 Therefore it is not completely surprising that activating mutations of RET cause increased production of epinephrine (discussed later in the section on MEN2-related pheochromocytoma).
The normal C cell synthesizes and secretes calcitonin, a peptide hormone important in the regulation of osteoclast function. Hyperplasia of C cells is associated with increased production and release of calcitonin and a change in the pattern of differential RNA processing that results in production of the alternative product, calcitonin gene–related peptide34 (reviewed in Chapter 57). It is not known whether increased calcitonin production results exclusively from the increased cell number or is related to increased expression of a cell-specific transcription factor that is a member of the helix-loop-helix family of transcription factors.35–37 Measurement of serum calcitonin basally and after a provocative test has been used to detect C cell hyperplasia.38,39 Carcinoembryonic antigen normally is produced by C cells40 and by MTC41; the serum concentration of carcinoembryonic antigen correlates roughly with tumor mass and provides an independent method of monitoring the mass of MTC.42 Chromogranin A43 and somatostatin44 are two other peptides frequently produced by normal C cells and by MTC. Neither is specific for MTC; therefore these two peptides have not been used extensively to diagnose or monitor patients with MTC.
The primary event in the development of neoplasia consists of clonal expansion of the C, adrenal medullary, and parathyroid cell populations. The initiating event in MEN2 and in approximately 25% of cases of sporadic MTC is an activating mutation of the RET proto-oncogene, a gene that encodes a tyrosine kinase receptor. Identification of these mutations and the RET-receptor system is a fascinating story that has relevance not only to MEN2 but also to several other genetic conditions that affect the nervous system.
The RET proto-oncogene was discovered in 1985 by Takahashi and coworkers.45,46 Several years later, Italian investigators discovered a transforming sequence in papillary thyroid carcinoma,47–49 a tumor derived from thyroid follicular cells, and showed it to be a naturally occurring rearrangement of RET. Subsequently, multiple different rearrangements have been identified in papillary thyroid carcinoma and are thought to be important in its genesis.50–52
Efforts to map the causative gene for MEN2 were initiated in the early 1980s and led to identification of a centromeric chromosome 10 locus in 1987.16,53 Subsequent mapping efforts narrowed the region to include the RET gene, and in 1993, Mulligan and coworkers discovered mutations of the RET proto-oncogene54—observations that were confirmed subsequently by Donis-Keller and others.55
The RET proto-oncogene encodes a tyrosine kinase receptor. Together with a second extracellular protein, GFRα-1 (glial cell–derived neurotrophic factor family receptor-α-1), it forms a receptor complex for glial cell–derived neurotrophic factor (GDNF),56 a small peptide discovered because of its ability to prevent neuronal cell death.57 The recognition that GDNF was a ligand for the RET/GFRα-1-receptor complex arose from a series of parallel but independent murine investigations in which the genes for RET58 and GDNF59–61 were deleted by homologous recombination techniques. The RET knockout mouse has a distinctive phenotype that includes failure of normal gastrointestinal neuronal (leading to a Hirschsprung-like phenotype) and kidney development. The recognition that GDNF was a ligand for RET grew out of the identification of a nearly identical phenotype in RET– and GDNF-deficient mice. The third component of this receptor system, GFRα-1,62 was identified by investigators seeking to identify a receptor for GDNF; the importance of this extracellular protein was verified subsequently by the finding of a phenotype in the GFRα-1 knockout identical to that found in the RET and GDNF knockouts.63,64
The RET/GFRα-1-receptor system normally is activated by GDNF.65 The GFRα-1 extracellular component is tethered to the plasma membrane and interfaces with RET to function as a receptor for GDNF. Interaction of GDNF with the receptor complex causes dimerization, autophosphorylation of a specific subset of tyrosine residues, and activation of ERK1/ERK2 (extracellular signal–regulated kinase) and JNK (c-Jun NH2-terminal kinase) downstream pathways66 (Fig. 151-2). Several discrete functions have been established for the receptor complex. The first is to cause normal neuronal and kidney development. The best understood is the interaction between the developing GDNF-expressing renal mesoderm and the ureteral bud where RET is expressed. Absence of GDNF or RET results in failure of ureteral bud invasion into the developing renal mesoderm,67 which leads to failure of collecting system development. Addition of a tiny pellet containing GDNF to the kidney derived from the RET-deficient mouse in vitro leads to normalization of ureteral branching.67 In the gastrointestinal tract, it is thought that the RET-expressing neural crest invades the developing gastrointestinal tract in response to temporal and spatial expression of GDNF. Failure of normal RET, GDNF, or GFRα-1 expression leads to a Hirschsprung phenotype.68 The second function of the receptor system is an antiapoptotic or anti–cell death effect.69 A component of the neuronal invasion of the gastrointestinal tract may be inhibition of neuronal cell death by this receptor system. Finally, growth-stimulatory functions of the receptor system are mediated through the tyrosine kinase receptor and its downstream signaling cascade. It is by activation of these growth-stimulatory or anti-apoptotic pathways that mutant RET is thought to cause neoplastic transformation in MEN2.70–72
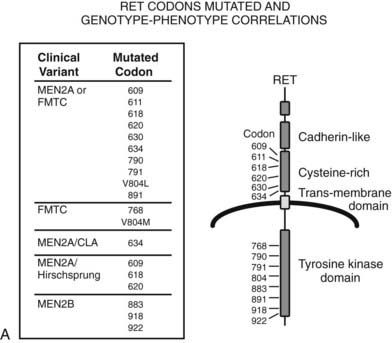
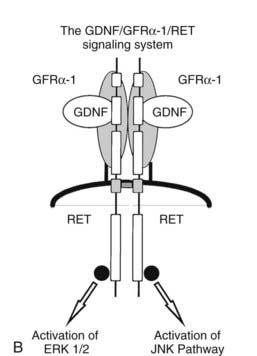
FIGURE 151-2. A, The RET proto-oncogene is a tyrosine kinase receptor (right). Germline mutations of both the extracellular (cysteine-rich) and intracellular (tyrosine kinase) domains cause hereditary medullary thyroid carcinoma (MTC). The clinical variants associated with germline mutations of specific codons are shown on the left. MEN2A, Multiple endocrine neoplasia type 2A; FMTC, familial MTC; MEN2A/CLA, MEN2A and cutaneous lichen amyloidosis variant; MEN2A/Hirschsprung, MEN2A and Hirschsprung’s disease; V804L, Val804Leu; and V804M, Val804Met. B, The glial cell–derived neurotrophic factor (GDNF)/glial cell–derived neurotrophic factor receptor-α (GFRα)/RET signaling system. GDNF binds to a receptor complex consisting of GFRα and RET. Autophosphorylation of RET results in activation of at least two distinct kinase pathways (ERK1/ERK2 [extracellular signal-regulated kinase] and JNK [c-Jun NH2-terminal kinase]). These pathways are linked to growth, differentiation, and antiapoptotic effects in cell types that express this complex.
Subsequent studies have demonstrated a total of four TGF-beta-like ligands (GDNF, artemin, persephin, neuturin) and at least four variants of GFRα (GFRα-1, GFRα-2, GFRα-3, and GFRα-4).73 Pairing of RET with one of the four GFRα variants creates specificity for one of the four TGF-β–like ligands. This receptor family has broad importance for normal neuronal, kidney, and neuroendocrine cell development.74 Important for understanding C cell biology is the recognition that persephin interacts with GFRα-4 and RET to determine the number of C cells and to regulate calcitonin production.22,75
The Clinical Syndromes
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2A
MEN2A is the association of MTC, pheochromocytoma, and hyperparathyroidism inherited as an autosomal dominant trait. Clinical features in the fully developed form of this syndrome include the presence of bilateral thyroid masses, manifestations of pheochromocytoma, and, less commonly, hyperparathyroidism (see Table 151-1). Other clinical features include diarrhea, renal stones, and the potential for sudden death related to elevated levels of catecholamines.
Medullary Thyroid Carcinoma
MTC generally is bilateral and multicentric and is set on a background of generalized C cell hyperplasia. Most commonly, the tumor appears as a chalky-white lesion within the upper portion of each lobe of the thyroid gland (see Fig. 151-1). The tumors frequently are multifocal, a finding thought to represent expansion of individual clones of cells rather than intrathyroidal metastasis62 (Fig. 151-3).
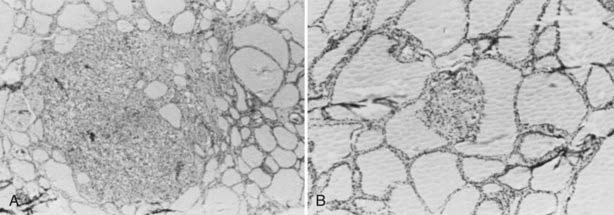
FIGURE 151-3. Histologic features of hereditary medullary thyroid carcinoma. A, Nodular C cell hyperplasia displacing an entire thyroid follicle. B, Microscopic medullary thyroid carcinoma.
The initiation of prospective screening with provocative calcium or pentagastrin tests in the early 1970s led to the identification of C cell hyperplasia as a precursor lesion for MTC.76 The progression from normal through hyperplasia, nodular hyperplasia, and microscopic and macroscopic carcinoma appears to occur over a number of years. Little is known about the age of initiation of clonal expansion, although C cell hyperplasia has been observed in gene carriers as young as 3 years. Metastasis has been described in patients with microscopic MTC.77
The biological behavior of MTC in MEN2A is variable. Metastasis to local lymph nodes occurs frequently with tumors larger than 1 cm. Although metastasis may be present, the tumor pursues a relatively indolent course in 80% of affected individuals. In about 5% to 10% of patients, the tumor pursues a more aggressive course, which can include early metastasis and death.78,79 Some kindreds have a familial pattern of tumor aggressiveness; in others, one or two members will display an aggressive pattern of tumor behavior against a general background of benignity. This characteristic has allowed MEN2A to remain undetected in some families over several generations, with the result that newly discovered kindreds continue to appear with some regularity.80–82 Characteristics of aggressive tumor behavior include early bone, pulmonary, or liver metastasis; loss of expression of the calcitonin gene; and a switch to production of calcitonin gene–related peptide, the alternatively produced product of the calcitonin gene. Distant metastasis occurs most commonly to liver, lung, and bone. Death usually is attributable to metastatic disease to liver and lung, although local growth of tumor can cause airway obstruction. The presence of metastatic disease in the liver usually is a poor prognostic feature with a median survival for hereditary MTC with distant metastasis of 4.3 years (M.D. Anderson Cancer Center, unpublished data), although several patients with hepatic metastasis have survived several decades.83,84
Diarrhea may be the initial complaint in patients with MTC. At first, the diarrhea may be a minor complaint, but with increasing tumor burden, the patient may have 10 to 20 stools per day. Unlike diarrhea associated with islet cell tumors, the stools usually are not voluminous.85 The cause of the diarrhea is unclear, although it is believed to be caused by a humoral factor produced by the tumor. Flushing has been observed in patients with MTC.
Another clinical finding associated with MTC is ectopic adrenocorticotropic hormone (ACTH) syndrome (see Chapter 15). The clinical syndrome is seen most frequently in patients with a large primary tumor or with metastatic MTC.86 Detailed analysis of MTC suggests that most express the pro-opiomelanocortin gene,87 but the peptide precursor is processed to produce ACTH in only a few tumors. The clinical features of hypercorticism may be subtle (muscle weakness, edema, and mild centripetal obesity). Clinicians should be aware of this clinical syndrome because patients with MTC and ectopic ACTH production may do well for long periods if the hypercorticism is controlled (see Chapter 15).
Pheochromocytoma
Clinically detectable pheochromocytoma will develop in approximately 50% of known gene carriers, although autopsy studies suggest that a larger percentage of gene carriers have abnormalities of the adrenal medullae.12,88 Generally, it is believed that the adrenal medullary cell, like the C cell, passes through a hyperplastic stage before the development of multicentric pheochromocytoma. It seems likely that the histologic abnormalities in the adrenal medulla occur in parallel with those observed in the C cell, although clinically apparent pheochromocytomas are only rarely detected before C cell abnormalities are diagnosed.
Unique Features of Pheochromocytomas Associated With MEN2A
Pheochromocytoma in MEN2A generally is limited to the adrenal gland. The occasional exception may be explained by the presence of ectopic adrenal rest tissue or rare neoplastic changes in nonadrenal chromaffin tissue, producing paraganglionomas.89 The pheochromocytomas usually are multicentric and are set on a background of diffuse adrenal medullary hyperplasia.12,88 Although malignant pheochromocytoma in MEN2A is rare,90–92 capsular invasion is observed frequently. No correlation has been found between capsular invasion and recurrence of the tumor.
Pheochromocytoma associated with MEN2A can be differentiated from sporadic pheochromocytoma by several unique features. The first is relative overproduction of epinephrine by the tumor. The earliest biochemical abnormality is an increase in plasma or urine epinephrine84,93–96 (Fig. 151-4). This may be caused by the positive regulation of PNMT by RET,31 which increases methylation of norepinephrine to produce epinephrine. A clinical observation that may be attributable to this biochemical finding is the relative lack of hypertension in patients with early pheochromocytomas and the predominance of β-adrenergic–like symptoms such as palpitations, tachycardia, and nervousness. In contrast, sporadic pheochromocytomas or those associated with von Hippel-Lindau syndrome are more commonly norepinephrine-producing tumors (see Fig. 151-4).96 It was shown that plasma metanephrine is a reliable indicator of pheochromocytoma in MEN2A, perhaps as sensitive as the measurement of plasma or urine catecholamines.96 Rarely, adrenal medullary hyperplasia may be associated with symptoms suggestive of pheochromocytoma but with few or no detectable abnormalities of catecholamines. Larger pheochromocytomas also are characterized by overproduction of norepinephrine, although the ratio of epinephrine to norepinephrine remains increased.84 Hypertension may be a clinical problem in patients with larger tumors. Sudden death related to pheochromocytoma occurred with some frequency in kindreds before routine prospective screening, and in some kindreds was as common a cause of death as MTC.
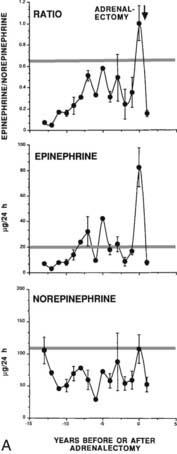
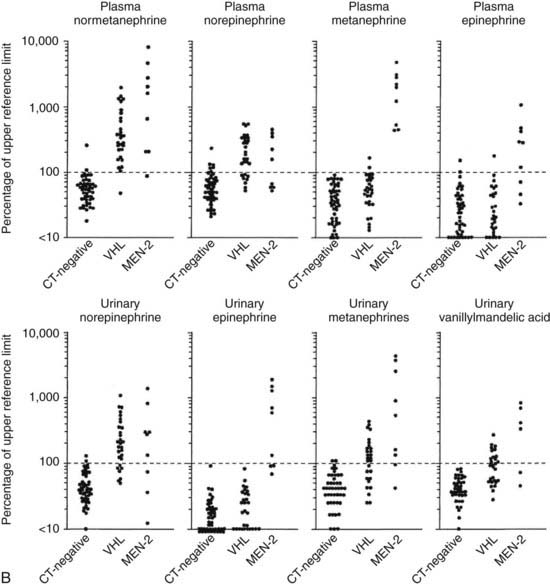
FIGURE 151-4. A, Mean 24-hour urinary norepinephrine and epinephrine excretion and the ratio of epinephrine-to-norepinephrine excretion in 11 prospectively screened patients subsequently proved to have pheochromocytoma. Each value is the mean of values in 11 prospectively screened patients before adrenalectomy. The horizontal line shows the upper limit of normal. To convert epinephrine values to nanomoles, multiply by 5.458; to convert norepinephrine values to nanomoles, multiply by 5.911. B, Plasma concentrations of normetanephrine, norepinephrine, metanephrine, and epinephrine (upper panels) and urinary excretion of norepinephrine, epinephrine, metanephrine, and vanillylmandelic acid (lower panels). Values are expressed as percentages of the upper reference limit for each test. Data on individual patients are shown for three groups of patients with von Hippel-Lindau disease and multiple endocrine neoplasia type 2 (MEN2) as follows: patients with von Hippel-Lindau disease or MEN2 in whom a pheochromocytoma was ruled out on the basis of normal computed tomography (CT-negative), patients with von Hippel-Lindau disease who had histologically verified pheochromocytomas (VHL), and patients with MEN2 who had histologically verified pheochromocytomas (MEN2). Values for patients with pheochromocytoma were determined when the tumors were first identified by CT. The dotted horizontal line represents the upper reference limit for each test. The scales are logarithmic.
(A, Data from Gagel RF, Tashjian AH Jr, Cummings T, et al: The clinical outcome of prospective screening for multiple endocrine neoplasia type 2A: an 18-year experience, N Engl J Med 318:478–484, 1988; B, Data from Eisenhofer G, Lenders JWM, Linehan WM, et al: Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2, N Engl J Med 340:1872–1879, 1999.)
Hyperparathyroidism
Hyperparathyroidism occurs in 15% to 20% of patients with the fully developed form of MEN2A. Parathyroid hyperplasia is the most common histologic abnormality; when parathyroid adenomas are found, they generally are set against a background of hyperplasia.7,8 The clinical features of hyperparathyroidism do not differ from those associated with sporadic hyperparathyroidism (see Chapter 56). Unlike MEN1, in which hyperparathyroidism generally is the first manifestation of the syndrome, hyperparathyroidism in MEN2 rarely occurs during the early phases of the syndrome. In addition, prospective screening of families with MEN2A led to the finding that hyperparathyroidism or early MTC did not develop in children who underwent thyroidectomy for C cell hyperplasia during a 10- to 15-year follow-up period.84 Whether the failure of hyperparathyroidism to develop in thyroidectomized children is related to partial removal of parathyroid tissue at the time of thyroid surgery and reflects an inadequate follow-up period or is related to removal of a growth stimulus by total thyroidectomy is unclear. In some families, hyperparathyroidism occurs early and is a prominent part of the syndrome.97
The MEN2A/Cutaneous Lichen Amyloidosis Variant
At least 20 kindreds have been identified in which MEN2A is associated with the development of a pruritic skin lesion over the upper part of the back98,99 (Fig. 151-5A, see Table 151-1). The first manifestation of the skin lesion is intermittent pruritus. The onset of pruritus generally precedes the development of a visible skin lesion by several years. The fully developed skin lesion has a lichenoid-papular appearance and may be unilateral or bilateral. Amyloid deposition analogous to that found in cutaneous lichen amyloidosis is found in more advanced lesions (Fig. 151-5B).
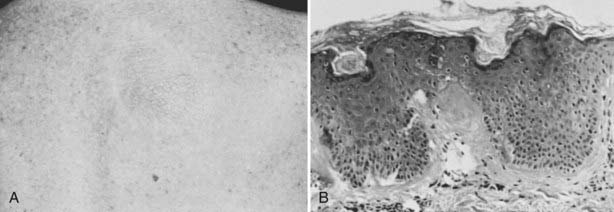
FIGURE 151-5. A, Characteristic cutaneous feature of lesion of cutaneous lichen amyloidosis. Patients with this lesion complain of intermittent pruritus and burning in the area of the skin lesion. B, Characteristic histologic features of cutaneous lichen amyloidosis showing deposition of amyloid at the interface between the dermis and the epidermis.
(From Gagel RF, Levy ML, Donovan DT, et al: Multiple endocrine neoplasia type 2a associated with cutaneous lichen amyloidosis, Ann Intern Med 111:802–806, 1989.)
Familial Medullary Thyroid Carcinoma
FMTC is another variant of MEN2A without other manifestations of MEN2A (see Table 151-1).100,101 The clinical characteristics of MTC in FMTC do not differ substantially from those of MEN2A, although the observation has been made that the FMTC variant is less aggressive. One viewpoint is that the FMTC-only syndrome is MEN2A but with a later onset of neoplasia, which makes it less likely that pheochromocytoma will develop during a normal lifetime.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2B
The association of MTC, pheochromocytoma, mucosal neuromas, a marfanoid body habitus, and the absence of hyperparathyroidism has been classified as MEN2B. Although it was described earlier, it was Williams and colleagues who compiled the several components into a distinct clinical syndrome.10 Most cases of this clinical syndrome are thought to represent new mutations because of failure to find evidence of disease in parents, although germline transmission of the disease with an autosomal dominant pattern of inheritance has been described in several kindreds.102–104
Medullary Thyroid Carcinoma and Pheochromocytoma in MEN2B
Development of MTC in MEN2B is thought to follow a pattern of progression similar to that described for MEN2A, but with a few differences. C cell hyperplasia and microscopic carcinoma, in general, develop much earlier in MEN2B. Children with metastatic MTC shortly after birth have been described.105,106 Death related to complications of metastatic MTC may occur before the third decade, although a larger experience that includes several multigenerational families suggests that the prognosis in an individual patient may be better than was previously considered.104,107 Pheochromocytomas in MEN2B occur in more than 50% of affected individuals and may develop at an early age. The clinical manifestations of pheochromocytoma do not differ substantially from those observed in MEN2A.
Mucosal Neuromas and Other Clinical Features of MEN2B
The most striking phenotypic feature of MEN2B is the presence of multiple mucosal neuromas. The presence of multiple neuromas located on the tongue tip, within the lips, and on the eyelids makes for a characteristic facies identifiable even in childhood108 (Fig. 151-6). Mucosal neuromas exist throughout the gastrointestinal tract (see Table 151-1). Gastrointestinal symptoms are the second most common reason for recognition of this syndrome.109 Children and adults frequently will have complaints of increased gas, abdominal pain, and, occasionally, obstruction or pseudo-obstruction caused by neuromatous tissue. Obstructive symptoms combined with diarrhea caused by MTC can produce a puzzling clinical syndrome characterized by alternating obstructive symptoms and diarrhea. It is important to exclude an anatomic cause of obstruction and, where possible, to debulk the mass of MTC to reduce diarrhea. Abdominal exploration generally is indicated only in patients with proven obstruction. Gastrointestinal neuromas associated with MEN2B can be confused with Hirschsprung’s disease, a condition also caused by mutations of the RET proto-oncogene.110
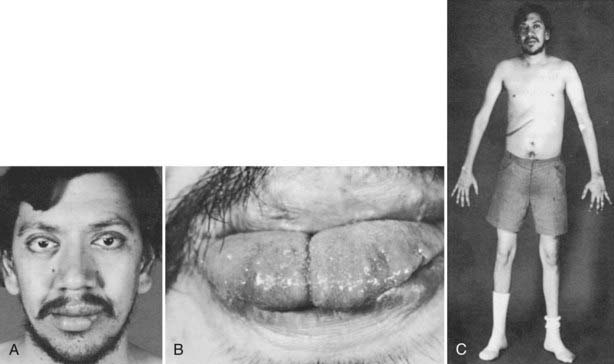
FIGURE 151-6. Characteristic facies with thickened lips and eyelids in a patient with multiple endocrine neoplasia (MEN) type 2B. Note that this patient had extensive reconstructive surgery on the left side of his lower lip (A) and the tip of his tongue (B) during childhood in an attempt to excise the neuromas. C, The marfanoid features of MEN2B, including the long arms, thin fingers, and altered body ratio. The abdominal scar resulted from removal of a unilateral pheochromocytoma.
Hyperparathyroidism is rare in MEN2B.111 Other clinical features frequently found in this syndrome include a marfanoid habitus with long, thin arms, an altered upper-to-lower body ratio, long fingers, hyperextensible joints, and slipped femoral epiphyses.112 The presence of these clinical features in Abraham Lincoln has led to the hypothesis that the sixteenth president of the United States of America may have suffered from MEN2B. Although no objective evidence of this association has been found, the marfanoid-like habitus and the presence of cutaneous neuroma-like features in Lincoln and two of his sons (Willie and Tad) and their early deaths lend some credibility to this argument.113
MEN2A and MEN2B have been viewed as two separate syndromes, but molecular studies have demonstrated that both occur as a result of mutations of the RET proto-oncogene. The identification of a mother with MEN2B and two children with MTC, one with the characteristic MEN2B phenotype and the second with no phenotypic features of MEN2B, suggests that the mucosal neuroma phenotype may not be 100% penetrant.114 Reports of corneal nerve thickening, a finding associated with MEN2B, in kindreds with MEN2A or FMTC,115 have provided additional evidence of overlap.
Screening for MEN2A
The primary goal of screening for MEN2 is to identify and treat the several manifestations of MEN2 before they become life threatening. A secondary goal is to provide genetic counseling to family members about the potential for transmission to the next generation. These goals become closely intertwined because family decisions regarding the next generation are based largely on experience in the current generation. The two life-threatening manifestations of MEN2 are metastasis from MTC and sudden death caused by pheochromocytoma. A 35- to 40-year experience with prospective screening now makes it reasonable to believe that death or serious morbidity caused by these manifestations can be prevented in most patients.
MEDULLARY THYROID CARCINOMA
Measurement of serum calcitonin basally and after a provocative test was the preferred method for diagnosis of MTC in MEN2 until genetic testing was introduced approximately 15 years ago; calcitonin assay remains a useful test for confirming the diagnosis and for following patients who have been treated. The sensitivity and specificity of the test are enhanced by the use of a provocative stimulus of calcitonin release, such as calcium,116 pentagastrin,117 or a combination of the two.118 The provocative stimulus results in a rapid (within 2 to 10 minutes) increase in serum calcitonin concentration.
Experience with pentagastrin (or combined calcium/pentagastrin) testing over the past 2 decades indicates that it is a reliable but not overly sensitive predictor of C cell abnormalities. The test is performed by injection of pentagastrin, 0.5 µg/kg, over a 5- to 10-second period, generally in the fasting state. The serum calcitonin level is measured basally and 2, 5, 10, and 15 minutes after the injection, although measurement of basal and 2- and 5-minute samples will reduce the cost of the test without affecting its sensitivity. The normal range is defined for each specific calcitonin assay: Normal values for immunoassays show pentagastrin-stimulated values less than 30 pg/mL for women and 110 pg/mL for men119; two-site immunoassays show stimulated values that generally are less than 5 pg/mL for female patients and less than 30 pg/mL for males.120 The major objections of patients to use of the test include several unpleasant side effects, including nausea, substernal tightness, flushing, tingling of the extremities, and the urge to void; these symptoms usually subside within 2 to 3 minutes of injection and are of variable severity. The patient should be warned of these side effects before the pentagastrin injection and should be reassured of their transient nature. No serious side effects have occurred when pentagastrin has been used for prospective screening in children. Anecdotal reports have described hypotension and flushing in patients with a significant tumor mass, and basal calcitonin measurements will suffice to assess tumor progression.42 Currently, pentagastrin is not manufactured in the United States, and calcium stimulation has replaced pentagastrin in situations in which a provocative test is still required.116
PHEOCHROMOCYTOMA
The goal of screening for pheochromocytoma is to identify and treat excess catecholamine production before the development of life-threatening manifestations. Experience over the past 2 decades indicates this can be accomplished by simple screening techniques repeated at regular intervals. A careful history is important and may provide the earliest clue to the presence of an adrenal medullary abnormality. Each suspected gene carrier should be queried annually about the presence of palpitations, nervousness or attacks of jitteriness, headaches, or other unusual vascular symptoms. Screening for excessive catecholamine production can be accomplished by annual measurement of plasma catecholamines and metanephrines. Urinary measurement of catecholamines and metanephrines is used less commonly today. The earliest indications of increased adrenal medullary mass include an increase in the 12- to 24-hour excretion of epinephrine and an increase in the ratio of epinephrine to norepinephrine in this collection (see Fig. 151-4A) or an elevated basal plasma epinephrine or metanephrine level (see Figure 151-4B).96,121 Measurement of vanillylmandelic acid is not useful for detection of pheochromocytoma.84
Screening for pheochromocytoma should begin by age 6 years. Adrenal medullary hyperplasia or pheochromocytoma has been described in the 10- to 12-year age range,84 and a 13-year-old child had hypertensive encephalopathy caused by a large pheochromocytoma.122 Parents should be instructed to be alert for symptoms of headache or jitteriness in children. Screening should be intensified during the child-bearing years, which also are the peak years for diagnosis of pheochromocytoma in this syndrome.
Several radiographic studies have been used for the diagnosis of pheochromocytoma in MEN2. Preoperative evaluation with computed tomography or magnetic resonance imaging is important for defining surgical anatomy and for determining bilaterality. Considerable controversy has developed over the use of 131I-labeled metaiodobenzylguanidine for the diagnosis of pheochromocytoma in MEN2.123 Although most investigators agree that this radioisotope is a very sensitive indicator of adrenal medullary hyperfunction, it may be too sensitive and its use may result in the detection of adrenal medullary hyperplasia (which is likely to be present in most gene carriers older than 20 years) before a significant increase in the production of catecholamines has occurred.124 This technique may be useful for identifying the rare extra-adrenal pheochromocytoma in a patient with catecholamine abnormalities and no identifiable adrenal abnormality.125 Arteriography is almost never indicated in the management of pheochromocytoma associated with MEN2 and should be performed only after adrenergic receptor antagonists have been administered (see Chapter 109).
HYPERPARATHYROIDISM
Screening for hyperparathyroidism in MEN2 is straightforward. Measurement of the serum calcium concentration every other year after age 10 years is adequate for early diagnosis.84,97,126 The finding of an elevated serum calcium level should prompt measurement of serum intact parathyroid hormone. Pheochromocytoma is a rare cause of hypercalcemia in MEN2 and should be excluded before parathyroid gland exploration is performed.84 Other, more common causes of hypercalcemia should be excluded by a careful history and physical examination and additional laboratory tests when appropriate (see Chapter 56).
Screening for MEN2B
Evidence indicates that MTC will develop in most patients with MEN2B diagnosed at an early age. Metastatic carcinoma has been described in children with MEN2B as young as 3 months. Because of this early expression of MTC, all agree that thyroidectomy should be performed at the earliest possible age in children with phenotypic features of MEN2B. It also is possible that the mucosal neuroma phenotype may not be 100% penetrant, thus making it mandatory to screen (through genetic analysis or calcitonin testing) all children born to a parent with MEN2B.113
Molecular Genetics
RET PROTO-ONCOGENE MUTATIONS IN MEN2A AND -2B
RET proto-oncogene mutations that cause MEN2 can be divided arbitrarily into extracellular and intracellular domain mutations.120 The most common mutations are those that change a cysteine in the extracellular domain to another amino acid. Mutations of several codons (292, 321, 533, 600, 603, 606, 609, 611, 618, 620, 630, 634, and 649) have been identified, and several other unusual mutations result in insertions or deletions in this region.127–129 Almost all these mutations cause receptor dimerization and autophosphorylation and lead to activation of downstream ERK1/ERK2 and JNK pathways.70–72 The most commonly mutated codon is 634, which accounts for almost 80% of mutations in MEN2.17 Mutations at codons 609, 611, 618, 620, and 630 account for approximately 15% of all mutations.55 Intracellular codons that are mutated in MEN2 include codons 666, 768,127,130 777, 781, 790, 791,131 804,130 844, 883,132–135 891,115 918,136 912,137 and 922.17 Fewer than 25 families each have been described for codons 666, 768, 777, 781,790, 791, 804, 844, 891, 912, and 922.137 The most common intracellular mutation is a Met918Thr coding change that is found in approximately 5% of all patients with MEN2.136 The codon 918 mutation causes receptor autophosphorylation in the absence of dimerization and activation of the ERK1/ERK2 and JNK pathways, as well as a different set of downstream substrate proteins.70,71,138
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


