FIGURE 150-1. Schematic representation of the distribution of 384 multiple endocrine neoplasia type 1 (MEN1) tumors in 220 patients with MEN1. The proportions of patients in whom parathyroid, pancreatic, or pituitary tumors occurred are shown in the respective boxes; for example, 94.5% of patients had a parathyroid tumor. The Venn diagram indicates the proportions of patients with each combination of tumors; for example, 37.7% (25.9% + 11.8%) of patients had both a parathyroid and a pancreatic tumor, whereas 2.3% of patients had a pancreatic tumor only. In addition to these tumors observed in one series,9 multiple facial angiofibromas were observed in 88% of 32 patients and collagenomas in 72% of these patients.55 The hormones secreted by each of these tumors are indicated: ACTH, Adrenocorticotropic hormone; GAS, gastrin; GCG, glucagon; GH, growth hormone; INS, insulin; NFT, nonfunctioning tumor; PRL, prolactin. Parathyroid tumors represent the most common form of MEN1 tumors and occur in approximately 95% of patients, with pancreatic islet cell tumors occurring in about 40% and anterior pituitary tumors occurring in around 30%.
(Adapted from Trump D, Farren B, Wooding C, et al: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) in 220 patients, Q J Med 89:653–669, 1996.)
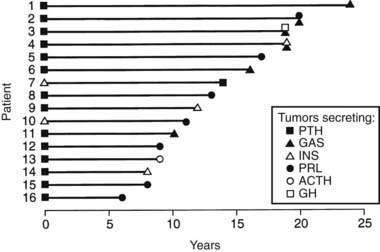
FIGURE 150-2. Order of tumor development in 16 patients with multiple endocrine neoplasia type 1 (MEN1). The interval between occurrence of the first tumor (time, 0 years) and subsequent tumors in each patient is shown. Parathyroid tumors were the first manifestation of MEN1 in 14 (88%) of 16 patients, and in the remaining 2 patients, insulinomas represented the first manifestation of MEN1. The interval for occurrence of the subsequent tumors ranged from 6 to 24 years in patients 16 and 1, respectively, and no correlation was found between the interval and tumor types. Abbreviations for the hormones secreted by each tumor are as indicated in Figure 150-1. PTH, Parathyroid hormone.
(From Trump D, Farren B, Wooding C, et al: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) in 220 patients, Q J Med 89:653–669, 1996.)
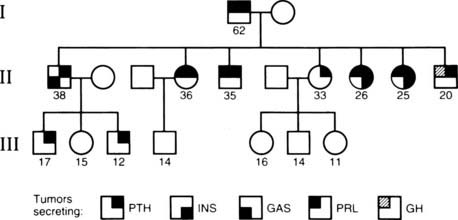
FIGURE 150-3. Variable expression of multiple endocrine neoplasia type 1 (MEN1) tumors within a family. In this family, the grandfather (generation I) had parathyroid tumors (PTH) and a prolactinoma (PRL). All of his seven affected children (generation II) had parathyroid tumors, and three daughters and one son also had prolactinomas, whereas another son had a somatotropinoma that secreted growth hormone (GH). In addition, two of his daughters had insulinomas (INS), and a son had gastrinomas (GAS). Two grandsons (generation III), who are 17 and 12 years old, have parathyroid tumors, which are the first manifestation of MEN1. Males (squares), females (circles), unaffected (open symbol), and affected (shaded quadrant) members are shown. The age in years is indicated below for each individual.
PARATHYROID TUMORS
Primary hyperparathyroidism is the most common feature of MEN1 and occurs in approximately 95% of all patients with MEN1.3,9,40,59–61,71–74 Patients may have asymptomatic hypercalcemia, nephrolithiasis, osteitis fibrosa cystica, vague symptoms associated with hypercalcemia (e.g., polyuria, polydipsia, constipation, or malaise), or occasionally peptic ulcers (see Chapter 62). Biochemical investigations reveal hypercalcemia, usually in association with raised circulating parathyroid hormone (PTH) concentrations. The hypercalcemia is usually mild, and severe hypercalcemia resulting in crisis or parathyroid cancers is a rare occurrence.58,59,75 Additional differences in the primary hyperparathyroidism of patients with MEN1, as opposed to those without MEN1, include an earlier age at onset (20 to 25 years versus 55 years) and an equal male/female ratio (1:1 versus 1:3). Primary hyperparathyroidism in patients with MEN1 is unusual before the age of 15 years, and the age of conversion from being unaffected to being affected has been observed to be between 20 and 21 years in some individuals.9
Surgical removal of the abnormally overactive parathyroids in patients with MEN1 is the definitive treatment, but it is controversial whether to perform subtotal or total parathyroidectomy and whether it should be performed at an early or late stage. Minimally invasive parathyroidectomy is not recommended,62 since all four parathyroid glands are usually affected with multiple adenomas or hyperplasia,59,62,76–80 although this histologic distinction may be difficult,81 and subtotal parathyroidectomy for primary hyperparathyroidism in patients with MEN1 has been associated with a high failure rate.78–80,82–85 Subtotal parathyroidectomy (i.e., removal of less than 3.5 glands) has resulted in persistent or recurrent hypercalcemia within 10 to 12 years after surgery in 40% to 60% of patients, and in hypocalcemia requiring long-term therapy with vitamin D or its active metabolite calcitriol in 10% to 30% of patients with MEN1.79,80,82–85 These rates are markedly higher than those observed for parathyroidectomies in patients who do not have MEN1, in whom recurrent hypercalcemia occurs in 4% to 16% and hypocalcemia in 1% to 8% of patients.82,86
To avoid neck reexploration, which is difficult, and to improve the treatment of primary hyperparathyroidism in patients with MEN1, total parathyroidectomy with autotransplantation of parathyroid tissue in the forearm has been performed.84,87–90 Both fresh and cryopreserved parathyroid tissue has been used for autotransplantation. The use of cryopreserved parathyroid tissue allows confirmation of hypoparathyroidism in the patient before autotransplantation, but unfortunately, only 50% of parathyroid grafts survive cryopreservation. The use of fresh parathyroid tissue for autotransplantation in the forearm results in viable grafts that secrete PTH. However, the presence of functioning autotransplanted parathyroid tissue leads to recurrent hypercalcemia in more than 50% of patients with MEN1, and surgical removal of transplanted grafts has been required. Thus, management of primary hyperparathyroidism in patients with MEN1 is difficult; parathyroid surgery in these patients is associated with a higher prevalence of persistent or recurrent hypercalcemia. Total parathyroidectomy, which would prevent such recurrence, has therefore been suggested as treatment of primary hyperparathyroidism in MEN1, with the resultant lifelong hypocalcemia being treated with oral calcitriol (1,25-dihydroxyvitamin D). However, the management of hypoparathyroidism can be challenging in some patients, even with the use of vitamin D and calcium replacement. One recommendation is that parathyroidectomy be reserved for symptomatic hypercalcemic patients with MEN1 and that asymptomatic hypercalcemic patients with MEN1 not have parathyroid surgery but have regular assessment for the onset of symptoms and complications, at which time total parathyroidectomy (or 3.5-gland parathyroidectomy), together with a possible transcervical near-total thymectomy, should be undertaken.84,85 The type of surgery (i.e., subtotal or total parathyroidectomy, with or without autotransplantation of parathyroid tissue) and its timing need careful consideration, and factors such as the surgical experience, the availability of facilities for long-term regular serum calcium monitoring, the accessibility of calcitriol (or vitamin D analogs), and the preferences of the patient should be taken into account.
PANCREATIC TUMORS
The incidence of pancreatic islet cell tumors (see Chapter 152) in patients with MEN1 varies from 30% to 80% in different series.9,27,59,76,91 Most of these tumors (see Table 150-1) produce excessive amounts of hormone (for example, gastrin, insulin, glucagon, or vasoactive intestinal polypeptide [VIP]) and are associated with distinct clinical syndromes, although some may remain nonfunctional or nonsecretory (see Fig. 150-1). These pancreatic islet cell tumors have an earlier age at onset in patients with MEN1 than in patients without MEN1.9
Gastrinoma
Zollinger and Ellison92 initially described two patients in whom non–β islet cell tumors of the pancreas were associated with recurrent peptic ulceration and marked gastric acid production, and gastrin was subsequently extracted from such tumors.93,94 The association of recurrent peptic ulceration, marked gastric acid production, and non–β islet cell tumors of the pancreas is referred to as the Zollinger-Ellison syndrome. Gastrin-secreting tumors (gastrinomas) represent more than 50% of all pancreatic islet cell tumors in patients with MEN19,59,60,72,73 (see Fig. 150-1), and approximately 20% of patients with gastrinomas will have MEN1.62,95 Gastrinomas, which may also occur in the duodenal mucosa,96 are the major cause of morbidity and mortality in patients with MEN1, and the prognosis is worse in patients with pancreatic primary tumors, metastases, ectopic Cushing’s syndrome, or markedly elevated plasma gastrin concentrations.97 Most MEN1 gastrinomas are malignant and will have metastasized in patients before a diagnosis is established.98,99 Gastrinomas occur more often in patients with MEN1 who are older than 40 years9 (Fig. 150-4), and recurrent, severe, multiple peptic ulcers (which may perforate) and cachexia are major contributors to the high mortality. Patients with Zollinger-Ellison syndrome may also suffer from diarrhea and steatorrhea. The diagnosis is established by demonstration of a raised fasting serum gastrin concentration in association with increased basal gastric acid secretion.100 Occasionally, intravenous provocative tests101 with either secretin (2 U/kg) or calcium infusion (4 mg Ca2/kg/h for 3 hours) are required to distinguish patients with Zollinger-Ellison syndrome from other patients with hypergastrinemia, such as those with antral G-cell hyperplasia. However, in patients with MEN1, Zollinger-Ellison syndrome does not appear to occur in the absence of primary hyperparathyroidism,60,102 and hypergastrinemia has also been reported to be associated with hypercalcemia.103 Thus, the diagnosis of Zollinger-Ellison syndrome may be difficult in some patients with MEN1. Moreover, successful treatment of the primary hyperparathyroidism with restoration of normocalcemia will significantly ameliorate the clinical symptoms and biochemical abnormalities in as many as 20% of patients with MEN1.85
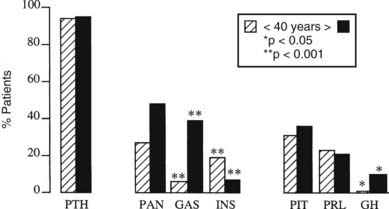
FIGURE 150-4. Development of 370 multiple endocrine neoplasia type 1 (MEN1) tumors before (hatched columns) and after (solid columns) 40 years of age in 220 affected individuals. The proportion of patients with parathyroid tumors (PTH), all pancreatic tumors (PAN), gastrinomas (GAS), insulinomas (INS), all pituitary tumors (PIT), prolactinomas (PRL), and somatotropinomas secreting growth hormone (GH) is shown for each group. Gastrinomas and somatotropinomas occurred more often in those older than 40 years, whereas insulinomas occurred more often in those younger than 40 years.
(Adapted from Trump D, Farren B, Wooding C et al: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) in 220 patients, Q J Med 89:653–669, 1996.)
Medical treatment of patients with MEN1 and Zollinger-Ellison syndrome is directed toward reducing basal acid output to less than 10 mmol/L, and such reduced acid output may be achieved by parietal cell H+-K+-adenosine triphosphatase (ATPase) inhibitors (e.g., omeprazole or lansoprazole), which have proved efficacious and become the drugs of choice for gastrinomas.104,105 Some patients may also require additional treatment with the histamine H2 receptor antagonists, cimetidine or ranitidine.105–107 The role of surgery in the treatment of gastrinomas in patients with MEN1 is controversial.99,105,108–113 The ideal treatment for a nonmetastatic gastrinoma situated in the pancreas is surgical excision of the gastrinoma.108,109 In addition, duodenal gastrinomas, which occur more frequently in patients with MEN1, have been treated successfully with surgery.96 However, in most patients with MEN1, gastrinomas are frequently multiple or extrapancreatic, and with the exception of duodenal gastrinomas, surgery has often not been successful.91,112–116 For example, the results of one study112 revealed that only 16% of patients with MEN1 were free of disease immediately after surgery, and at 5 years, this number had decreased to 6%. The respective outcomes in patients without MEN1 were better, at 45% and 40%. Given these findings, the majority of physicians and about 50% of surgeons recommended a nonsurgical management for gastrinomas in MEN1.62 The use of tumor-localization studies involving ultrasonography, endoscopic ultrasonography, computed tomography (CT), nuclear magnetic resonance imaging (MRI), selective abdominal angiography, venous sampling, or somatostatin-receptor scintigraphy demonstrated that these techniques may sometimes help to improve the surgical success rate.112,117–122 For example, transhepatic selective venous gastrin sampling in one study demonstrated that patients with MEN1 had either diffuse gastrin secretion from multiple pancreatic sites or localized gastrin secretion from a single region.117 The combined use of intraarterial calcium injections with hepatic venous gastrin sampling has also been shown to help determine the location of gastrinomas.122 The patients in whom gastrin secretion was localized benefited from resection of the gastrinoma by partial pancreatectomy and required no drug therapy postoperatively. Surgery for gastrinomas that are larger than 2 to 2.5 cm has been recommended, insofar as the disease-related survival in these patients was shown to be improved following surgery.123 Total gastrectomy is rarely undertaken now and would perhaps be considered only for persistently noncompliant patients.100 Treatment of disseminated gastrinomas is difficult, and chemotherapy with streptozotocin and 5-fluorouracil,91,124 hormonal therapy with octreotide or lanreotide (which are human somatostatin analogs),125 hepatic artery embolization,126 administration of human leukocyte interferon,92,127 and removal of all resectable tumor128 have all been successful occasionally.
Insulinoma
Insulinomas, β islet cell tumors that secrete insulin, represent 10% to 30% of all pancreatic tumors (see Fig. 150-1) in patients with MEN1.3–9,61,76,91 Insulinomas also occur in association with gastrinomas in 10%9,61,129,130 of patients with MEN1, and the two tumors may arise at different times. Insulinomas occur more often in patients with MEN1 who are younger than 40 years (see Fig. 150-4), and many of them arise in individuals younger than 20 years,9 whereas in patients without MEN1, insulinomas generally occur in those older than 40 years.3,9 Insulinomas may be the first manifestation of MEN1 in 10% of patients (see Fig. 150-2), and approximately 4% of patients with insulinomas will have MEN1.95 Patients with an insulinoma present with hypoglycemic symptoms that develop after a fast or exertion and improve after glucose intake (see Chapters 47 and 152). The most reliable test is a supervised 72-hour fast, and biochemical investigations reveal increased plasma insulin concentrations in association with hypoglycemia.131 Circulating concentrations of C peptide and proinsulin, which are also increased, are useful in establishing the diagnosis. It also is important to demonstrate an absence of sulfonylureas in the plasma and urine samples obtained during the investigation of the hypoglycemia. Medical treatment, which consists of frequent carbohydrate meals and diazoxide or octreotide, is not always successful; surgery is the optimal treatment. Most insulinomas are multiple and small, and preoperative localization with endoscopic ultrasonography, CT scanning, or celiac axis angiography, preoperative and perioperative percutaneous transhepatic portal venous sampling, selective intraarterial stimulation with hepatic venous sampling, and intraoperative direct pancreatic ultrasonography have been undertaken to improve the success rate of surgery.93,121,132–136 Surgical treatment, which ranges from enucleation of a single tumor to a distal pancreatectomy or partial pancreatectomy, has been curative in many patients.89,108,132 Chemotherapy consisting of streptozotocin, 5-flurouracil, and doxorubicin or hepatic artery embolization has been used for metastatic disease.125,126,137
Glucagonoma
Glucagonomas—β islet cell, glucagon-secreting, pancreatic tumors—occur in fewer than 3% of patients with MEN19,69,138–143 (see Fig. 150-1). The characteristic clinical manifestations of a skin rash (necrolytic migratory erythema), weight loss, anemia, and stomatitis may be absent, and the presence of the tumor may have been detected in an asymptomatic patient with MEN1 undergoing pancreatic imaging or detected by glucose intolerance and hyperglucagonemia9,143 (see Chapter 152). The tail of the pancreas is the most frequent site for glucagonomas, and surgical removal is the treatment of choice. However, treatment may be difficult because approximately 50% to 80% of patients have metastases at the time of diagnosis.142,143 Medical treatment with somatostatin analogs (e.g., octreotide or lanreotide) or chemotherapy with streptozotocin and 5-fluorouracil or dimethyltriazeno-imidazole carboxamide (DITC) has been successful in some patients, and hepatic artery embolization has been used to treat metastatic disease.144,145
VIPoma
Patients with vasoactive intestinal peptide (VIP)omas, which are VIP-secreting pancreatic tumors, develop watery diarrhea, hypokalemia, and achlorhydria (see Chapter 152). This clinical syndrome has been referred to as the Verner-Morrison syndrome,146 the WDHA syndrome,147 or the VIPoma syndrome.148 VIPomas have been reported in only a few patients with MEN1,60,146,149–151 and the diagnosis is established by excluding laxative and diuretic abuse, confirming a stool volume in excess of 0.5 to 1.0 L/day during a fast, and documenting a markedly increased plasma VIP concentration. Surgical management of VIPomas, which are mostly located in the tail of the pancreas, has been curative. However, in patients with unresectable tumor, treatment with somatostatin analogs such as octreotide and lanreotide,125,151 streptozotocin with 5-fluorouracil,152 corticosteroids,153 indomethacin,154 metoclopramide,155 and lithium carbonate156 has proved beneficial, and hepatic artery embolization has been useful for the treatment of metastases.
PPoma
Pancreatic polypeptide (PP) secreting tumors, which secrete pancreatic polypeptide, are found in a large number of patients with MEN1.57,157,158 No pathologic sequelae of excessive PP secretion are apparent, and the clinical significance of PP is unknown, although the use of serum PP measurements has been suggested for the detection of pancreatic tumors in patients with MEN1.158,159 Many PPomas may have been unrecognized or classified as nonfunctioning tumors113,160 (see Fig. 150-1). The management of these nonfunctioning pancreatic islet cell tumors in the asymptomatic patient is controversial.62 One recommendation is to undertake surgery irrespective of tumor size, once an unequivocal biochemical diagnosis is made. Another recommendation is to undertake surgery if the imaged tumor is 1 cm or larger,99,101 whereas yet another recommendation is to undertake surgery only if the tumor is larger than 3 cm or is growing.110 Pancreatoduodenal surgery may be successful in removing the tumors in 80% of patients, but more than 40% of patients will develop complications that include diabetes mellitus, frequent steatorrhea, early and late dumping syndromes, and other gastrointestinal symptoms.111 However, between 47% and 60% of patients were alive at a follow-up period of 5 years or more, and global quality of life (QOL) assessments in the surgically treated patients was similar to that in the reference population.111,113 These studies suggest that early diagnosis and surgical removal of pancreatic endocrine tumors in MEN1 patients prolongs survival, without significant comprise in patient-perceived QOL.111,113 When considering these recommendations, it is important to consider that occult metastatic disease (i.e., tumors not detected by imaging investigations) is likely to be present in a substantial proportion of these patients at the time of presentation.109
Somatostatinoma
Somatostatin, which inhibits secretion of growth hormone and many other hormones, has been demonstrated to be present in the gastrointestinal tract, particularly in the pancreatic islets.161–163 Pancreatic tumors secreting somatostatin are associated with the somatostatinoma syndrome, which is characterized by hyperglycemia, cholelithiasis, low acid output, steatorrhea, diarrhea, abdominal pain, anemia, and weight loss.164,165 Although 7% of pancreatic islet cell tumors in MEN1 secrete somatostatin, the somatostatinoma syndrome does not appear to have been reported in any patient with MEN1, a finding that may reflect the inhibitory action of somatostatin on endocrine cell proliferation and secretion.
GHRHoma
GHRHomas, or tumors that secrete growth hormone-releasing hormone (GHRH), have been reported in some patients with MEN1,166,167 and it is estimated that approximately 33% of patients with GHRHomas will have other MEN1-related tumors. GHRHomas may be diagnosed by finding elevated serum concentrations of growth hormone and GHRH. More than 50% of GHRHomas occur in the lung, 30% occur in the pancreas, and 10% are found in the small intestine.167,168
PITUITARY TUMORS
The incidence of pituitary tumors in patients with MEN1 varies from 15% to 90% in different series,9,27,56,59,77 and about two-thirds of these are microadenomas (diameter < 1 cm). Approximately 60% of MEN1-associated pituitary tumors secrete prolactin (see Fig. 150-1), fewer than 25% secrete growth hormone, 5% secrete adrenocorticotrophic hormone (ACTH), and the remainder appear to be nonfunctioning, with some secreting glycoprotein subunits.9,53,72,95,169–175 However, pituitary tumors from MEN1 patients may show immunoreactivity to several hormones, and in particular there is a higher occurrence of somato-lactotropinomas.175 Prolactinomas may be the first manifestation of MEN1 in fewer than 10% of patients, and somatotrophinomas occur more often in patients older than 40 years9 (see Fig. 150-4). Fewer than 3% of patients with anterior pituitary tumors will have MEN1.9,53–55 The clinical manifestations of these tumors in patients with MEN1 are similar to those in patients without MEN1 and depend on the hormone secreted and the size of the pituitary tumor: patients may have symptoms of hyperprolactinemia (e.g., amenorrhea, infertility, and galactorrhea in women and impotence in men) or have acromegaly or Cushing’s disease. In addition, enlarging pituitary tumors may compress adjacent structures such as the optic chiasm or normal pituitary tissue and cause bitemporal hemianopia or hypopituitarism, respectively. Treatment of pituitary tumors in patients with MEN1 is similar to that in patients without MEN1 and consists of medical therapy or selective hypophysectomy by the transsphenoidal approach if feasible, with radiotherapy being reserved for residual unresectable tumor.
ASSOCIATED TUMORS
Patients with MEN1 may have tumors involving tissues other than the parathyroids, pancreas, and pituitary. Carcinoid, adrenal cortical tumors, facial angiofibromas, collagenomas, thyroid tumors, and lipomatous tumors (see Fig. 150-1) have been described in association with MEN1.2,3,9
Carcinoid Tumors
Carcinoid tumors, which occur in more than 3% of patients with MEN1 (see Fig. 150-1), may be inherited as an autosomal-dominant trait in association with MEN1.9,170,176 The carcinoid tumor may be located in the bronchi,177 the gastrointestinal tract,177–181 the pancreas,182 or the thymus.170,183,184 Bronchial carcinoids in patients with MEN1 occur predominantly in women (male/female ratio, 1 : 4), whereas thymic carcinoids occur predominantly in men, with cigarette smokers having a higher risk for these tumors.183,184 The course of thymic carcinoids in MEN1 appears to be particularly aggressive, and CT or MRI is recommended for the early detection of thymic and bronchial carcinoids.62 Most patients are asymptomatic and do not have the flushing attacks and dyspnea associated with the carcinoid syndrome, which usually develops after the tumor has become malignant and metastasized to the liver.185 Gastric carcinoids, of which the type II gastric enterochromaffin-like (ECL)-cell carcinoids (ECLomas) are associated with MEN1 and the Zollinger-Ellison syndrome, may be detected incidentally at the time of gastric endoscopy for dyspeptic symptoms in MEN1 patients.185–187 These tumors, which may be found in over 70% of MEN1 patients, are usually multiple and smaller than 1.5 cm.188,189 Little is known about their malignant potential,185 but treatment with somatostatin analogs, such as octreotide or lanreotide, has resulted in regression of these ECLomas.190
Adrenocortical Tumors
The incidence of asymptomatic adrenocortical tumors in patients with MEN1 has been reported to be as high as 40%.27,76,138,191–193 Most of these tumors, which may include cortical adenomas, hyperplasia, multiple adenomas, nodular hyperplasia, cysts, or carcinomas, are nonfunctioning.9,138,191–193 However, functioning adrenocortical tumors in patients with MEN1 have been documented to cause hypercortisolemia and Cushing’s syndrome,9,138,191 as well as primary hyperaldosteronism, as in Conn’s syndrome.194–197
Lipomas
Subcutaneous lipomas may occur in more than 33% of patients with MEN1,27,179 and frequently they are multiple. In addition, visceral, pleural, or retroperitoneal lipomas may occur in patients with MEN1.
Facial Angiofibromas and Collagenomas
Studies of patients with MEN1 have revealed that multiple facial angiofibromas occur in 22% to 88% of those studied and collagenomas in 0% to 72%.198,199 MEN1 angiofibromas were clinically and histologically identical to those observed in patients with tuberous sclerosis, with the exception that in patients with MEN1, angiofibromas were also present on the upper lip and vermilion border of the lip, which are areas not involved in tuberous sclerosis. These cutaneous findings, which occur with a higher frequency in patients with MEN1, may provide a useful means for possible presymptomatic diagnosis198,199 of MEN1 in the relatives of a patient with MEN1. However, it is important to note that combined occurrence of MEN1 and tuberous sclerosis has been observed.200
Thyroid Tumors
Thyroid tumors consisting of adenomas, colloid goiters, and carcinomas have been reported to occur in more than 25% of patients with MEN1.27,76 However, the prevalence of thyroid disorders in the general population is high, and it has been suggested that the association of thyroid abnormalities in patients with MEN1 may be incidental and not significant.
Molecular Genetics
MODELS OF TUMOR DEVELOPMENT
The development of tumors may be associated with mutations or inappropriate expression of specific normal cellular genes, which are referred to as proto-oncogenes.2,37,201–203 Two types of oncogenes, dominant and recessive, have been described. Activation of dominant oncogenes leads to transformation of the cells containing them, and examples include the chromosomal translocations associated with the occurrence of chronic myeloid leukemia204,205 and Burkitt’s lymphoma.206 In these conditions, mutations that lead to activation of the oncogene are dominant at the cellular level, and therefore only one copy of the mutated gene is required for the phenotypic effect. Such dominantly acting oncogenes may be assayed in cell culture by first transferring them into recipient cells and then scoring the numbers of transformed colonies, a technique referred to as the transfection assay. However, in some inherited neoplasms such as retinoblastoma, which may also arise sporadically,207 tumor development is associated with two recessive mutations that inactivate oncogenes, and these mutations are referred to as recessive oncogenes. In the inherited tumors, the first of the two recessive mutations is inherited via the germ cell line and is present in all the cells. This recessive mutation is not expressed until a second mutation, within a somatic cell, causes loss of the normal dominant allele. The mutations causing the inherited and sporadic tumors are similar, but the cell types in which they occur are different. In inherited tumors, the first mutation occurs in the germ cell, whereas in sporadic tumors, both mutations occur in the somatic cell. Thus, the risk of tumor development in an individual who has not inherited the first germline mutation is much smaller because both mutational events must coincide in the same somatic cell. In addition, the apparent paradox that the inherited cancer syndromes are due to recessive mutations but dominantly inherited at the level of the family may be explained: In individuals who have inherited the first recessive mutation, loss of the single remaining wild-type allele is almost certain to occur in at least one of the large number of cells in the target tissue. This cell will be detected because it forms a tumor, and almost all individuals who have inherited the germline mutation will express the disease, even though they inherited a single copy of the recessive gene.1,202 This model involving two (or more) mutations in the development of tumors is known as the “two-hit” or Knudson’s hypothesis.208,209 The normal function of these recessive oncogenes appears to be regulation of cell growth and differentiation, and these genes have also been referred to as antioncogenes, tumor-suppressor genes, or gatekeeper genes.37,201–203 An important feature that has facilitated investigation of these genetic abnormalities is that loss of the remaining allele (i.e., the “second hit”) often involves large-scale loss of chromosomal material.2,34 This second hit may be detected by comparing DNA sequence polymorphisms in leukocytes and tumor obtained from a patient and observing loss of heterozygosity (LOH) in the tumor (Fig. 150-5).
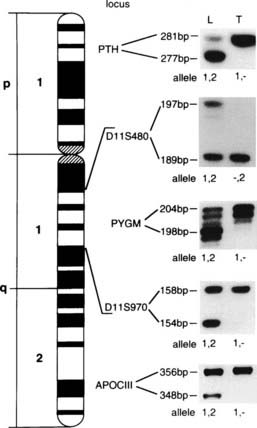
FIGURE 150-5. Loss of heterozygosity (LOH) involving polymorphic loci from chromosome 11 in a parathyroid tumor from a patient with familial multiple endocrine neoplasia type 1 (MEN1). The microsatellite polymorphisms obtained from the patient’s leukocyte (L) and parathyroid tumor (T) DNA at the PTH, D11S480, PYGM, D11S970, and APOCIII loci are shown. These microsatellite polymorphisms have been identified with specific primers for each of the loci that have been localized to chromosome 11 and are shown juxtaposed to their regions of origin on the short (p) and long (q) arms of chromosome 11. The microsatellite polymorphisms are assigned alleles (see Fig. 150-6). For example, D11S480 yielded a 197-bp product (allele 1) and a 189-bp product (allele 2) after polymerase chain reaction amplification of leukocyte DNA, but the tumor cells have lost the 197-bp product (allele 1) and are hemizygous (allele −,2). Similar losses of alleles are detected by using the other DNA markers, and an extensive loss of alleles involving the whole of chromosome 11 is observed in the parathyroid tumor of this patient with MEN1. In addition, the complete absence of bands suggest that this abnormality has occurred within all the tumor cells studied and indicates a monoclonal origin for this MEN1 parathyroid tumor.
(From Pang JT, Thakker RV: Multiple endocrine neoplasia type 1, Eur J Cancer 30:1961–1968, 1994.)
IDENTIFICATION OF THE MEN1 GENE AND MUTATIONS
The gene causing MEN1 was localized to chromosome 11q13 by genetic mapping studies that investigated MEN1-associated tumors for LOH (see Fig. 150-5) and by segregation studies in families with MEN138–40,210,211 (Fig. 150-6). The results of these studies, which were consistent with Knudson’s model for tumor development,210,211 indicated that the MEN1 gene represented a putative tumor-suppressor gene.38–40 Further genetic mapping studies defined a region of less than 300 kb as the minimal critical segment that contained the MEN1 gene, and characterization of genes from this region led to identification in 1997 of the MEN1 gene,42,43,212,213 which consists of 10 exons with a coding region of 1830 bp (Fig. 150-7) that encodes a novel 610–amino acid protein referred to as menin.42 The minimal promoter region is located within a few hundred bp upstream of exon 2, and a region further upstream, which contains a series of cis-regulatory sequences, modulates the activity of this minimal promoter, whose expression also depends on the cell type.214,215 The main transcript of the MEN1 gene is a 2.8-kb mRNA. However, at least six alternative transcripts have been reported, with variations in their content of the 5′-untranslated region; none of these affect the coding region.216 One additional very rare variant that would result in an elongation of the reading frame by 5 bases at the exon 2/intron 2 junction has also been reported.217 Menin is ubiquitously expressed and is predominantly a nuclear protein in nondividing cells.218 Analysis of the minimal promoter region of the MEN1 gene revealed that it did not contain a consensus TATA box for transcriptional initiation. However, binding sites for the SP1 transcriptional factor and nuclear factor (NF) 1 are located in both promoter regions, together with a recognition site for nuclear factor κB (NF-κB).214,215 Overexpression of menin in an inducible cell-culture system decreased MEN1 promoter activity, whereas down-regulation of MEN1 expression by RNA interference resulted in a compensatory activation of promoter activity.214 Thus, expression of the MEN1 gene is regulated by a feedback mechanism that involves its own protein product, menin.214,215,217
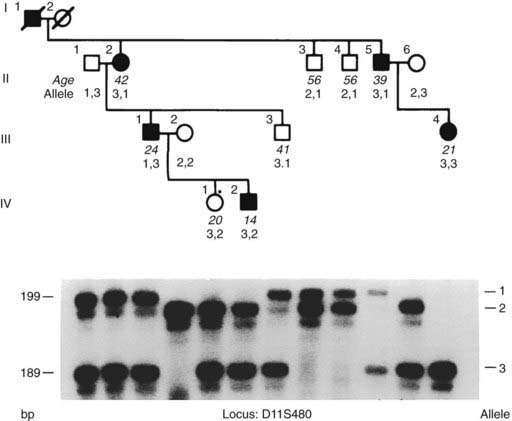
FIGURE 150-6. Segregation of D11S480, a polymorphic locus from chromosome 11q13 (see Fig. 150-5), and multiple endocrine neoplasia type 1 (MEN1) in a family. Genomic DNA from the family members (upper panel) was used with γ[32P]-adenosine triphosphate (ATP) for polymerase chain reaction (PCR) amplification of the polymorphic repetitive element (CA)n at this locus.2,34 The PCR amplification products were separated on a polyacrylamide gel and detected by autoradiography (lower panel); the products ranged in size from 189 to 199 bp. Alleles were designated for each PCR product and are indicated on the right. For example, individuals II.1, II.2, and III.1 have two pairs of bands on autoradiography. The upper pair of bands is designated allele 1 and the lower pair of bands is designated allele 3; these three individuals are therefore heterozygous (alleles 1, 3). A pair of bands for each allele is frequently observed in the PCR detection of microsatellite repeats. The upper band in the pair is the “true” allele, and the lower band in the pair is its associated “shadow,” which results from slipped-strand mispairing during PCR. Segregation of these bands and their respective alleles together with the disease can be studied in the family members whose alleles and ages are shown. In some individuals, inheritance of paternal and maternal alleles can be ascertained; the paternal allele is shown on the left. Individuals are represented as unaffected male (open square), affected male (solid square), unaffected female (open circle), and affected female (solid circle). Individual II.2 is affected and heterozygous (alleles 3, 1), and examination of her affected child (III.1), grandchild (IV.2), sibling (II.5), and niece (III.4) reveals inheritance of allele 3 with the disease. The unaffected individuals II.3, II.4, and III.3 have not inherited this allele 3. However, the daughter (IV.1) of individual III.1 has inherited allele 3 but remains unaffected at age 20 years; this may be either a representation of age-related penetrance (see Fig. 150-9) or a recombination between the disease and D11S480 loci. Thus, in this family, the disease and D11S480 loci are cosegregating in eight of the nine children, but in one individual (IV.1), assuming 100% penetrance (see later) in early childhood, recombination is observed. Thus, MEN1 and D11S480 are cosegregating in eight of nine of the meioses and not segregating in one of nine meioses, and the likelihood that the two loci are linked at θ = 0.11, that is, 11% recombination, is (8/9)8 × (1/9)1. If the disease and the D11S480 loci were not linked, the disease would be associated with allele 1 in half (1/2) of the children and with allele 3 in the remaining half (1/2) of the children, and the likelihood that the two loci are not linked is (1/2)9. Thus, the odds ratio in favor of linkage between the MEN1 and D11S480 loci at θ = 0.11 in this family is (8/9)8 × (1/9)1 ÷ (1/2)9 = 22.17 : 1, and the LOD score (i.e., log10 of the odds ratio favoring linkage) = 1.34 (i.e., log10 22.17). A LOD score of +3, which indicates a probability in favor of linkage of 1000 : 1, establishes linkage. LOD scores from individual families also can be summated, and such studies revealed that the peak LOD score between the MEN1 and DS11S480 loci was greater than +3, thereby establishing linkage between the MEN1 and D11S480 loci.319
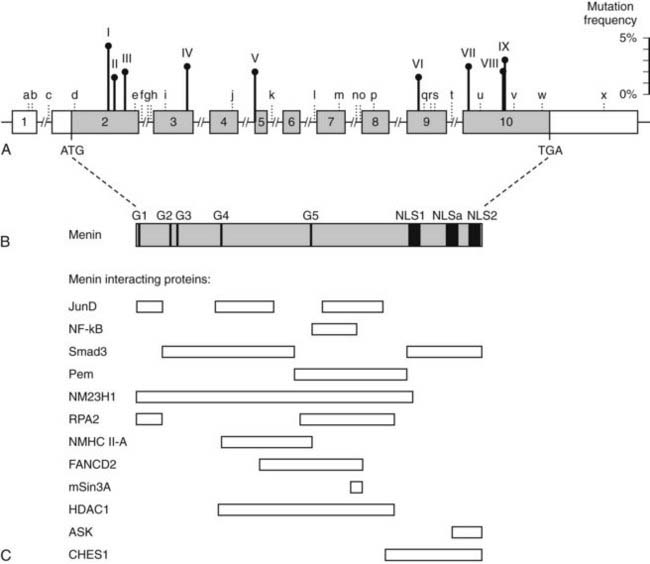
FIGURE 150-7. Schematic representation of the genomic organization of the MEN1 gene, its encoded protein (menin) and regions that interact with other proteins. A, The human MEN1 gene consists of 10 exons that span more than 9 kb of genomic DNA and encodes a 610-amino-acid protein. The 1.83-kb coding region (shaded region) is organized into 9 exons (exons 2 to 10) and 8 introns (indicated by a line but not to scale). The sizes of the exons (boxes) range from 41 to 1297 bp, and that of the introns range from 80 to 1564 bp. The start (ATG) and stop (TGA) codons in exons 2 and 10, respectively, are indicated. Exon 1, the 5′ part of exon 2, and the 3′ part of exon 10 are untranslated (open boxes). The promoter region is located within a few 100 bp upstream of exon 2. The sites of the nine germline mutations (I to IX) that occur with a frequency >1.5% (Table 150-2) are shown, and their respective frequencies (scale on the right) are indicated by the vertical lines above the gene. These germline mutations, which collectively represent 20.6% of all reported germline mutations, are: I- c.249_252delGTCT; II- c.292C>T; III- c.358_360delAAG; IV4- c.628_631delACAG; V- c.784-9G>A; VI- c.1243C>T; VII- c.1378C>T; VIII- c.1546delC; IX- c.1546_1547insC. The locations of the 24 polymorphisms (a-x, Table 150-4) are illustrated. B, Menin has three nuclear localization signals (NLSs) at codons 479–497 (NLS1), 546–572 (NLSa), and 588–608 (NLS2), indicated by closed boxes, and five putative guanosine triphosphatase (GTPase) sites (G1-G5) indicated by closed bars. C, Menin regions that have been implicated in the binding to different interacting proteins (Table 150-5) are indicated by open boxes. These are JunD (codons 1–40, 139–242, 323–428); nuclear factor κB (NF-κB) (codons 305–381); Smad3 (codons 40–278, 477–610); placenta and embryonic expression, Pem (codons 278–476); NM23H1 (codons 1–486); a subunit of replication protein A (RPA2) (codons 1–40, 286–448); NMHC II-A (codons 154–306); FANCD2 (codons 219–395); mSin3A (codons 371–387); HDAC1 (codons 145–450); ASK (codons 558–610) and CHES1 (codons 428–610). The regions of menin that interact with GFAP, vimentin, Smad 1/5, Runx2, MLL-histone methyltransferase complex and estrogen receptor α (ERα) remain to be determined.
(From Lemos MC, Thakker RV: Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene, Hum Mutat 29:22–32, 2008.)
Mutations of the MEN1 gene (see Fig. 150-7; Fig. 150-8) have been characterized, and a total of 1336 mutations (1133 germline and 203 somatic mutations) have been reported in the first decade following the identification of the gene.217 The 1133 germline mutations of the MEN1 gene, which consist of 459 different mutations, are scattered throughout the entire 1830-bp coding region and splice sites of the MEN1 gene42,43,66,217,219–222 (see Fig. 150-7). Approximately 23% are nonsense mutations, 41% are frameshift deletions or insertions, 6% are in-frame deletions or insertions, 9% are splice-site mutations, 20% are missense mutations, and 1% are whole or partial gene deletions (Fig. 150-9).217 More than 10% of the MEN1 mutations arise de novo and may be transmitted to subsequent generations.66,213,217 It also is important to note that between 5% and 10% of patients with MEN1 may not harbor mutations in the coding region of the MEN1 gene,42,43,66,217,219,221,222 and that these individuals may have whole gene deletions223 or mutations in the promoter or untranslated regions which remain to be identified. One study showed that approximately 33% of patients who do not have mutations within the coding region have large deletions involving complete exons.224 Such abnormalities will not be easily detected by DNA sequence analysis.
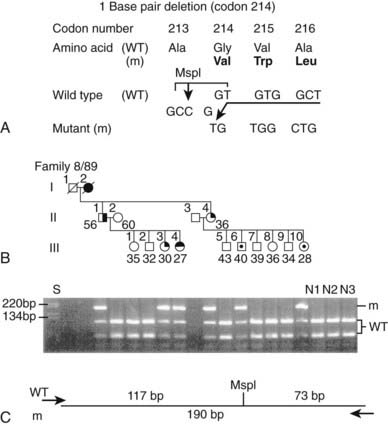
FIGURE 150-8. Detection of mutation in exon 3 in family 8/89 by restriction enzyme analysis. DNA sequence analysis of individual II.1 revealed a 1-bp deletion at the second position (GGT) of codon 214 (A). The deletion has caused a frameshift that continues to codon 223 before a stop codon (TGA) is encountered in the new frame. The 1-bp deletion results in the loss of an MspI restriction enzyme site (C/CGG) from the normal (wild-type, WT) sequence (A), and this finding has facilitated detection of this mutation in the other affected members (II.4, III.3, and III.4) of this family (B). The mutant (m) polymerase chain reaction product is 190 bp, whereas the WT products are 117 and 73 bp (C). The affected individuals were heterozygous, and the unaffected members were homozygous for the WT sequence. Individuals III.6 and III.10, who are 40 and 28 years old, respectively, are mutant gene carriers who are clinically and biochemically normal because of the age-related penetrance of this disorder (see Fig. 150-13B). These individuals would still require screening (see Fig. 150-13A) by clinical, biochemical, and radiologic assessment because they still have a residual risk (i.e., 100% – age-related penetrance) of 2% and >13%, respectively, of tumors developing by age 60 years. Individuals are represented as male (square); female (circle); unaffected (open); affected with parathyroid tumors (solid upper right quadrant), gastrinoma (solid lower right quadrant), or prolactinoma (solid upper left quadrant); and unaffected mutant gene carriers (dot in the middle of the open symbol). Individual I.2, who is dead but was known to be affected (tumor details not known), is shown as a solid symbol. The age is indicated below for each individual at diagnosis or at the time of the last biochemical screening. The standard size marker (S) in the form of the 1-kb ladder is indicated. Cosegregation of this mutation with MEN1 in family 8/89 and its absence in 110 alleles from 55 unrelated normal individuals (N1-3 shown) indicate that it is not a common DNA sequence polymorphism.
(Adapted from Bassett JHD, Forbes SA, Pannett AAJ, et al: Characterisation of mutations in patients with multiple endocrine neoplasia type 1 (MEN1), Am J Hum Genet 62:232–244, 1998.)
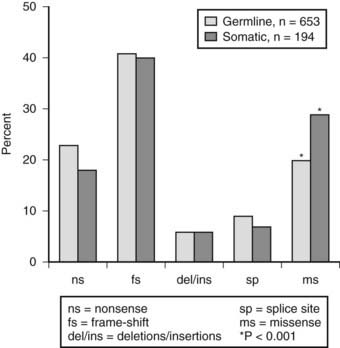
FIGURE 150-9. Frequency of germline and somatic MEN1 mutations. A total of 1133 germline mutations and 203 somatic mutations have been reported,219 and these are of diverse types (e.g., nonsense, frameshifts, deletions, insertions, splice-site, and missense mutations). The frequencies of each type of mutation in the germline and somatic groups are similar, with the exception of the missense mutations, which are found more frequently in tumors (i.e., the somatic group).219
Most (75%) of the MEN1 mutations are inactivating217 and are consistent with those expected in a tumor-suppressor gene.202 The mutations not only are diverse in their types but are also scattered (see Fig. 150-7) throughout the 1830-bp coding region of the MEN1 gene, with no evidence for clustering as observed in MEN212 (see Table 150-1). However, some of the mutations have been observed to occur several times in unrelated families (see Fig. 150-7 and Table 150-2). Mutations at nine sites in the MEN1 gene accounted for over 20% of all the germline mutations (see Table 150-2). Of these nine types of mutations, five are deletional and insertional mutations involving codons 83 and 84 (nt359 del 4), 120 (Lys[K]120 del), 210 to 211 (nt 738 del 4) (Fig. 150-10), and codons 514 to 516 (nt 1656-7 del or ins C); one is a novel acceptor splice site in intron 4, and three are nonsense mutations (Arg98Stop, Arg415Stop, and Arg460Stop).217,222 These mutations at these nine different sites could be considered to represent potential “hot spots” (see Tables 150-1 and 150-2). Such deletional and insertional hot spots may be associated with DNA sequence repeats, which may consist of long tracts of either single nucleotides or shorter elements ranging from dinucleotides to octanucleotides.66 The DNA sequences in the vicinity of codons 83 and 84 in exon 2, and codons 210 to 211 (see Fig. 150-10) in exon 3, contain CT and CA dinucleotide repeats, respectively, flanking the 4-bp deletions; this finding would be consistent with a replication-slippage model in which misalignment of the dinucleotide repeat takes place during replication, followed by excision of the 4-bp single-stranded loop.66 A similar replication-slippage model may also be involved at codons 119 and 120, each of which consists of AAG nucleotides encoding a lysine (K) residue. The deletions and insertions of codon 516 involve a poly(C)7 tract, and a slipped-strand mispairing model also is the most likely mechanism to be associated with this mutational hot spot.66,217 Thus, the MEN1 gene appears to contain DNA sequences that may render it susceptible to deletional and insertional mutations.
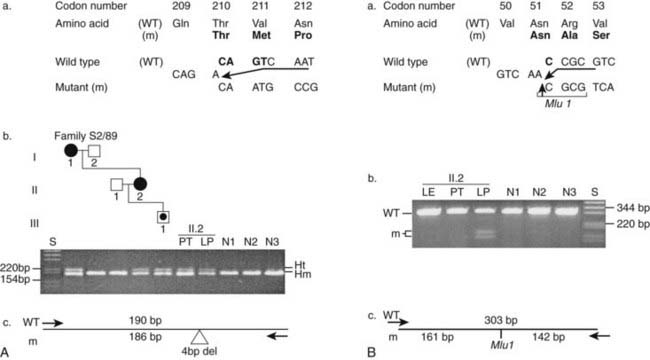
FIGURE 150-10. The detection of germline and somatic mutations in a lipoma from a multiple endocrine neoplasia type 1 (MEN1) patient. DNA sequence analysis of the lipoma (LP) from individual II.2 revealed two mutations, one germline (A) and one somatic (B). A, The germline mutation consisted of a 4-bp deletion (CAGT) involving codons 210 and 211, which is one of the nine frequently occurring mutations (see Table 150-2). The mutation is predicted to result in a frameshift with the introduction of 11 missense amino acids followed by a premature stop at codon 222 (a). After polymerase chain reaction (PCR) amplification of the mutation, the PCR products result in the formation of homoduplexes (Hm) and heteroduplexes (Ht), which facilitate the detection of the mutation in the family members and in a parathyroid tumor (PT) and lipoma (LP) from individual II.2 by agarose gel electrophoresis (b and c). The absence of this 4-bp deletion in 110 alleles from 55 unrelated normal individuals (N1-N3 shown) established that this was not a common sequence polymorphism. B, The somatic mutation in the lipoma (LP) from individual II.2 consisted of a 1bp deletion (C) at codon 51. The mutation resulted in the gain of an MluI restriction enzyme site, which facilitated the detection of the mutation (b and c) and the demonstration of its absence in the leukocytes (LE) and parathyroid tumor (PT) from individual II.2, and in 55 normal individuals (N1-N3 shown). After PCR amplification and MluI digestion, a 303-bp product was obtained for the wild-type (WT) allele, whereas two products of 161-bp and 142-bp were obtained for the mutant (m) alleles, which were resolvable by agarose gel electrophoresis (b and c). Individual I.1 had parathyroid tumors and a gastrinoma; II.2 had parathyroid tumors, prolactinoma, and lipoma; and individual III.1, who is 18 years old, is asymptomatic and biochemically normal, represents a carrier (dot in the middle of symbol) for the MEN1 mutation. There is an age-related penetrance for MEN1 (see Fig. 150-13). Squares, Males; circles, females; open symbols, unaffected; solid symbols, affected.
(From Pannett AA, Thakker RV: Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis, J Clin Endocrinol Metab 86(9):4371–4374, 2001.)
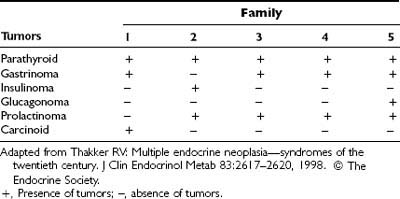
Correlations between MEN1 mutations and clinical manifestations of the disorder appear to be absent. For example, a detailed study of five unrelated families with the same 4-bp deletion in codons 210 and 211 (Table 150-3) revealed a wide range of MEN1-associated tumors20,66,217; all affected family members had parathyroid tumors, but members of families 1, 3, 4, and 5 had gastrinomas, whereas members of family 2 had insulinomas. In addition, prolactinomas occurred in members of families 2, 3, 4, and 5 but not in family 1, which was affected with carcinoid tumors. Another study of seven unrelated families with the same g→a novel acceptor splice-site mutation in intron 4 revealed a similarly wide range of MEN1-associated tumors and a lack of genotype/phenotype correlation.222 The apparent lack of genotype/phenotype correlation, which contrasts with the situation in MEN2 (see Table 150-1),12 together with the wide diversity of mutations in the 1830-bp coding region of the MEN1 gene, makes mutational analysis for diagnostic purposes in MEN1 time consuming and expensive.20
Table 150-3. Multiple Endocrine Neoplasia Type 1–Associated Tumors in Five Unrelated Families With a 4-Base-Pair Deletion at Codons 210 and 211
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


