RET STRUCTURE AND FUNCTION
The RET gene consists of over 20 exons spanning a minimum of 30 kilobases of genomic DNA. The protein product of the gene encodes a cell surface receptor. The gene consists of an extracellular domain (exons 1 to 10), a transmembrane domain (exon 11), and an intracellular domain (exons 12 onwards) (Fig. 90-1).12
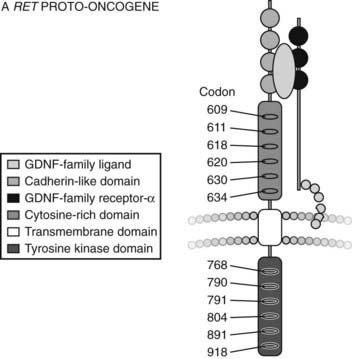
FIGURE 90-1. Structure of RET Proto-oncogene.
(From Cote GJ, Gagel RF: Lessons learned from the management of a rare genetic cancer. N Engl J Med 349:1566–1568, 2003.)
The RET protein is membrane-bound, with its intracellular portion having tyrosine kinase activity. The extracellular domain extends for the initial 635 amino acids and includes an area of cadherin homology and a conserved cysteine-rich region immediately adjacent to the transmembrane domain.13 The cysteine-rich domain is typical of the transforming growth factor beta (TGF-β) superfamily.14 The protein is glycosylated to produce receptors with molecular weights of 150 and 170 kilodaltons. The 170-kD form is present in the cell membrane, while the 150-kD form is an immature form present in the cell cytoplasm.15 Phosphorylation of RET tyrosine residues results in activation of several intracellular second messenger systems and signaling pathways.16 These include phospholipase Cγ/protein kinase C (PLCγ/PKC), c-Jun N-terminal kinases (c-Jun/JNK), products of the proto-oncogene Src-related kinases, and nuclear factor κB (NF-κB). Other downstream targets include the Ras/Raf/ERK (extracellular regulated kinase) and phosphatidylinositol-3-kinase (P13-kinase)/Akt pathways. Through these signaling cascades, RET appears to have a central role in cellular proliferation, differentiation, and migration.17–19 Interestingly, in addition to its role in endocrine neoplasia, aberrant RET signaling has been associated with distinct phenotypes. Hirschsprung’s disease occurring with MEN2 is described in subjects with codon 609, 618, and 620 mutations,20 and cutaneous lichen amyloidosis with codon 634 mutations.21
RET LIGANDS
There was intense interest in the early 1990s in identifying the ligand for RET. In 1996 the first ligand, glial cell line–derived neurotrophic factor (GDNF), was identified.22 Other ligands were soon identified, including neurturin (NTN),23 artemin,24 and persephin.25
RET activation by ligand appears to occur via membrane-bound proteins that function as the ligand-binding domain of a ligand/receptor complex. The link between RET and its first ligand, GDNF, was suggested by the study of mice that were deficient in GDNF expression. These mice were very similar to RET knockout mice, with absent or limited development of the kidney and an absence of enteric neurons in the colon and small intestine.26
GDNF-family ligands bind to specific GDNF-family receptor proteins, all of which form receptor complexes and signal through the RET receptor, tyrosine kinase. Additional proteins, GDNF receptor alpha (GFRα) or GDNF-family receptor alpha 1 (GFRα-1), were found to be essential for the high-affinity binding of GDNF to RET and for its consequent functional effects.27 A single molecule of ligand binds two GFRα-1 molecules, which interact with two molecules of RET and lead to activation of RET tyrosine kinase. Similarly, the other ligands such as neurturin, persephin, and artemin bind to different GFRαs before eventual receptor activation occurs, with consequent effects. It was suggested that MEN2 families with no identifiable RET mutations may have GDNF mutations, but so far no such mutations have been described in MEN2. However, somatic GDNF mutations have been found in sporadic pheochromocytomas.28
FUNCTIONAL EFFECTS OF RET MUTATIONS
Although the genotypes do overlap, in general, different codons are affected in either MEN2A or MEN2B. These mutations in different regions of the RET proto-oncogene increase the transforming activity of the kinase by distinct mechanisms. In MEN2A, the extracellular cysteine-rich or transmembrane domains are predominantly affected. Such mutations lead to a loss of cysteine residues. As a result, two RET molecules may undergo ligand-independent dimerization, with resultant constitutive activation of tyrosine kinase activity.29 A mutation in codon 634, usually a cysteine-to-arginine substitution (TGC to CGC), occurs in 85% of cases of MEN2A.17 In contrast, MEN2B is usually caused by a mutation in the intracellular tyrosine kinase domain. Codon 918 is involved in 95% of cases, in a methionine-to-threonine substitution (ATG to ACG).17 This alters the kinase specificity, with alternative substrate phosphorylation and altered downstream signaling.30 The rarer intracellular RET mutations in codons 768 and 804 have been shown to produce “gain of function” of the receptor, although the precise mechanism by which tumor formation occurs is unclear.31
Histogenesis
C CELLS
The calcitonin-secreting C cells of the thyroid originate from the neural crest. They are located mainly in the upper and middle thirds of the thyroid gland. They are members of the amine-precursor uptake and decarboxylation (APUD) family and express a variety of neuroendocrine markers, including calcitonin, chromogranin A, neuron-specific enolase, and serotonin.32 Calcitonin is a very effective tumor marker for the monitoring of MTC. Carcinoembryonic antigen (CEA) is also a C-cell marker used in clinical practice. C cells usually display an endocrine phenotype but may revert to a neuronal phenotype following malignant transformation.32 The alternately spliced calcitonin gene-related peptide (CGRP) is then the predominant protein expressed by the cells.
The biological role of C cells is not precisely clear. They are involved in calcium homeostasis, and they contribute to the thyroid follicle’s microenvironment. Pharmacologically, calcitonin inhibits calcium resorption from bone, and this has been used therapeutically for treatment of Paget’s disease, postmenopausal bone loss, and hypercalcemia.
C-CELL HYPERPLASIA
C-cell hyperplasia (CCH) is the pathologic description of an increase in C-cell numbers that can precede malignancy. Criteria for the diagnosis of CCH are variable in the literature, although consensus suggests that there should be more than 50 C cells per field at ×100 magnification.33 C cells may increase in number with age, and CCH is a common finding in normal thyroids at autopsy.34 CCH is no longer considered to be a specific marker of familial MTC. Multifocal CCH is regarded as a precursor lesion to hereditary MTC. Its progression to microscopic MTC is variable and may take many years.35
Clinical Relevance of Germline RET Mutations in MEN2A, FMTC, and MEN2B
MEN2A AND FMTC
Extensive studies of MEN2A families from around the world have now clarified that 97% will have a germline RET mutation, usually in the cysteine-encoding codons 609, 611, 618, 620, or 634. The codon 634 mutation in exon 10 predominates, usually with an arginine substitution, although other substitutions such as by tyrosine or glutamine are described.10 In FMTC, 88% of families have a germline mutation in one of the listed cysteine codons or, more rarely, in intracellular codons including 768, 790, and 791. The common MEN2A C634R mutation of cysteine to arginine (TGC to CGC) is not found in FMTC.36 RET mutations in the intracellular exons 13, 14, and 15 are less common in MEN2A, although they will be detected by routine sequencing of these exons. Isolated cases of kindreds with germline mutations in other exons (including exons 5 and 8) have been reported, and these may account for cases of apparently mutation-negative FMTC.17
MEN2B
In 95% of MEN2B families, the germline RET mutation at codon 918 in exon 16 occurs, causing mutation of methionine to threonine (ATG to ACG).37 This mutation is in the tyrosine kinase domain and is specific to MEN2B.9 Of note, this codon 918 RET mutation is also found somatically in the tumor tissue in some sporadic MTCs. A germline RET codon 918 mutation can also occur de novo, where MEN2B patients have no family history of the disease. However, they still have a 50% chance of passing this mutation on to their offspring.38 Rarer tyrosine kinase domain mutations occur in codons 88339 and 891.12
Specific Genotype/Phenotype Correlations
There has been much interest in the prediction of phenotypic behavior from genotype analysis. As published by the International RET Consortium, the presence of any codon 634 RET mutation correlates significantly with the presence of both pheochromocytoma and hyperparathyroidism. One study suggested more aggressive behavior of MTC with the common arginine substitution at codon 634 (C634R), compared to the C634Y mutation.40 The C634R mutation has not been associated with FMTC. It has been suggested that FMTC is a milder variant of MEN2A, and that pheochromocytoma may eventually occur if the patient survives long enough.10 In support of this concept, it had previously been believed that the codon 804 mutation was specific to FMTC. Subsequent reports have described pheochromocytoma occurring in these families, albeit at an older age than in other MEN2A kindreds.41,42 The occurrence of different phenotypes in different families with the same germline RET mutation suggest a role for more complex gene interactions. The rarer phenotypes associated with MEN2 include Hirschsprung’s disease with RET codon 609, 618, and 620 mutations43 and cutaneous lichen amyloidosis with codon 634 mutations.44 The codon 918 mutation is MEN2B specific.17
Clinical Presentation of MTC and MEN2
Patients with MTC may present with a palpable thyroid lump or with the symptoms of calcitonin excess—in particular, diarrhea and flushing. Diarrhea does not usually occur until the calcitonin level is very high (at least 10-fold above the upper limit of the normal range). Unfortunately it is very likely that the MTC will have spread to regional lymph nodes by the time diarrhea occurs. Familial cases of MTC will have different presentations depending on the specific RET mutation, as described earlier. Patients with MEN2B will have a more aggressive phenotype and may present in early childhood with diarrhea from very high calcitonin levels or with the other features of the MEN2B syndrome.1 Mucosal neuromas can occur on the tongue, in the lips, and on the eyelids, giving a distinctive facial appearance. Neuromas in the gastrointestinal tract can cause abdominal pain, gaseous distension, and even pseudo-obstruction. Hirschsprung’s disease can have a similar presentation in children and is due to inactivating mutations in the extracellular domain of RET, as distinct from the activating mutations of the tyrosine kinase domain seen in MEN2B patients.1 MEN2B patients may also have a marfanoid body habitus and corneal nerve thickening. Some patients with MEN2A will also show corneal nerve thickening. Pheochromocytoma occurs in 50% of MEN2B patients, sometimes at a young age, but hyperparathyroidism is very rare.
The purpose of biochemical and genetic screening in MEN2 families is to detect MTC well before it becomes clinically apparent, because once clinically evident, it may have spread to lymph nodes.
Screening in MEN2 and FMTC
BIOCHEMICAL SCREENING
The pentagastrin (PG) stimulation test was used for many years to identify biochemically affected members of MEN2 families. The advent of genetic screening has dramatically reduced the need for this test, although it still has a role in follow-up and management. A number of limitations of the PG test have been uncovered since RET mutation analysis became available. It is possible for RET mutation-negative members of MEN2 families to have elevated calcitonin responses to pentagastrin stimulation.45
In the early literature, some of these positive PG tests led to thyroidectomy, and C-cell hyperplasia was found in some patients but not in others.46 This is probably because CCH can be a normal variant.34 PG testing can also be used to detect hereditary MTC carriers in families where no RET mutation is identified, although these families are now rare. Adverse effects from the PG test include retrosternal discomfort, nausea, vomiting, metallic taste, abdominal cramps, esophageal spasm, and tachycardia.
GENETIC SCREENING
RET mutations are clustered tightly, and the analysis can be confined to specific exons. This compares favorably with genetic analysis in MEN1, where the mutations are scattered. Once a mutation is identified, other family members are screened with a straightforward analysis for that specific mutation. Various techniques have been used, and currently, direct polymerase chain reaction (PCR)-based sequencing is performed in most laboratories. Other methods include restriction enzyme digestion of PCR products47 with subsequent sequencing,48 single-strand conformation polymorphism, and denaturing gradient gel electrophoresis.49 Denaturing high-performance liquid chromatography can also be used to identify mutations, provided that appropriate positive controls are available, since there are multiple polymorphisms in the RET gene in normal individuals.50 Linkage analysis is not commonly performed, although it may rarely have a role if no RET mutation can be identified with other techniques.51 Testing should be performed on two separate occasions on two different blood samples to avoid the risk of sample mix-ups. RET mutation-negative members of known MEN2 families (i.e., where RET mutation is identified in the family) can be reassured that their risk of developing MTC is no greater than for the general population.
Genetic screening must therefore be comprehensive and include sequencing of exons 10, 11, 13, 14, 15, and 16.52
Management Implications of Genetic Results
TIMING OF PROPHYLACTIC THYROIDECTOMY
The advent of genetic screening has allowed prophylactic thyroidectomy to be performed earlier than it would be by traditional biochemical screening. Logically, this would be expected to lead to an improvement in the prognosis of RET-mutation carriers. Long-term data is slowly accumulating to support the practice of prophylactic thyroidectomy based on genetic screening rather than on clinical or biochemical screening. Consensus was reached at the MEN ’97 Workshop that the decision to perform thyroidectomy in MEN2 should be based predominantly on the result of RET-mutation testing rather than on calcitonin testing.53
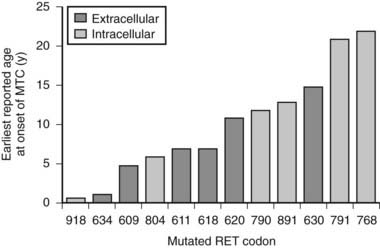
The specific mutated RET codon may be correlated with the degree of aggressiveness of the MTC. A three-level risk stratification system has been adopted since its inception at the Seventh International Workshop on Multiple Endocrine Neoplasia in 1999.52 Category 1, the highest risk group, includes all MEN2B-mutation carriers, because mutations in RET codon 883 and 918 are associated with the most aggressive MTC. Such patients should have thyroidectomy within the first 6 months of life because metastases within the first year of life have been described. Those with RET codon 611, 618, 620, or 634 are classified as category 2, having a high risk of MTC, and thyroidectomy before 5 years of age is recommended. MTC with the codon 634 mutation has been reported as early as age 2 years, although cases of MTC are generally rare before the age of 5 years in MEN2A and FMTC. Thyroidectomy at this age is technically possible without significant morbidity. Category 3 includes those with RET codon 609, 768, 790, 791, 804, and 891 mutations, having the lowest risk among the stratification categories. It should be noted that a single case of metastatic MTC in a 6-year-old was reported in a family with the codon 804 RET mutation.54 Some have recommended thyroidectomy by age 5 years, whereas others have suggested it by age 10 years, for gene-positive members of category 3–risk families. If baseline calcitonin is elevated, then thyroidectomy should be performed immediately, regardless of the RET mutation found. Histology in this setting is likely to show MTC rather than C-cell hyperplasia, with the associated risk of lymph node spread.55 Fig. 90-2 describes the earliest age of reported MTC for each of the known RET mutations. Early prophylactic surgery also alleviates the need for ongoing unpleasant PG stimulation testing being performed in a child.
STRATEGY AND EXTENT OF THYROIDECTOMY
Although there has been a general consensus regarding the timing of prophylactic thyroidectomy based on risk stratification, the strategy of central lymph node dissection at prophylactic thyroidectomy is controversial. Most advocate thyroid surgery with central node dissection for MEN2B. The practice of routine central-node clearance is most important in asymptomatic gene carriers with the higher-risk mutations but can be modified for lower-risk mutations with normal preoperative calcitonin.
The Role of Somatic RET Mutations
SPORADIC MTC
Somatic mutations of RET are observed in a proportion of sporadic MTC cases. The commonest somatic RET mutation is the codon 918 ATG-to-ACG (M918T) mutation. Its prevalence varies widely (from 28% to 86%) in different series.56 This may reflect either geographic differences in population studies or perhaps methodological differences. Other somatic mutations reported in sporadic MTC include codon 883 in exon 15 and codon 634 in exon 11.57 Although not a universal finding, smaller series had suggested that the somatic RET codon 918 mutation was associated with a poorer prognosis in sporadic MTC, correlating the mutation with distant metastases and tumor recurrence.58,59,60 A recent large study of 100 sporadic MTC patients with a 10-year follow-up period supported this concept.57 This study reported increased lymph node metastases at diagnosis, increased likelihood of persistent disease, and lower overall survival in those subjects with a somatic RET mutation.57
SOMATIC MUTATIONS IN HEREDITARY MTC
Somatic RET mutations have also been described in hereditary MTC. The somatic codon 918 RET mutation has been reported, albeit rarely, in association with germline RET mutations in exons 10 or 11.61 This important observation negated the theory that the codon 918 somatic RET mutation could be used as a specific marker of sporadic disease. Others have observed the somatic codon 918 RET mutation occurring in patients with a germline codon 768 RET mutation.62 Microdissection techniques have uncovered the fact that somatic RET mutations may be found in some regions of an MTC tumor but not in others.63 It has been suggested that the presence of a somatic mutation in addition to the germline RET mutation provides a “second hit” that may explain the more aggressive behavior of some familial cases of MTC.
ROUTINE GERMLINE SCREENING OF SPORADIC MTCS
It is now recommended that all new cases of sporadic MTC should have germline screening for RET mutations. Unsuspected familial cases are uncovered in approximately 6% of such patients despite an apparently negative family history.64 This was confirmed in a recent series of apparently sporadic MTC, in which 7.3% of subjects were found to possess a germline mutation. Lower-risk RET mutations in the non-cysteine codons were most commonly discovered, including codons 804, 891, and 768, although cysteine codon mutations were also found. The majority of these unsuspected familial cases were diagnosed as FMTC, based on the absence of other endocrine neoplasia at follow up.65 Of note, genetic screening of first-degree relatives should be undertaken if a germline RET mutation is identified in a subject with MTC.
Given that more than 97% of MEN2 families have identifiable RET mutations, a negative-germline RET-mutation test provides a high degree of certainty that a particular individual has sporadic MTC. This is assuming the genetic analysis is comprehensive. It is important to include exons 13, 14, and 15 because mutations in these exons are most likely to manifest late, with a lower prevalence of pheochromocytoma, and are more likely to escape detection as familial.66
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


