FIGURE 33-1. A human islet of Langerhans stained by immunofluoresence for insulin (green) and glucagon (blue).


FIGURE 33-2. A, Paul Langerhans (shown here in a rare family photograph) died young from tuberculosis. B, The face page of the thesis of Paul Langerhans defended on February 18, 1869.
(From Schadewaldt H: Geschichte des diabetes mellitus. Berlin, Springer-Verlag, 1975, pp 52–53.)
Islet Structure in Health
In humans, approximately one million islets of Langerhans are scattered in the pancreas.26–29 Islets vary greatly in size, with larger islets providing most of the insulin-secreting β cells and typically containing ≈2000 β cells.26,27,30 Each islet has its own complex anatomy, with the core consisting mainly of β cells that are tightly interconnected by gap junctions31–33 (Fig. 33-3), surrounded by a mantle of other endocrine cells, including glucagon-secreting α cells, somatostatin-secreting δ cells, and pancreatic polypeptide–secreting (PP) cells.29,34–36 In humans, ≈35% of islet cells are nonendocrine cells. The nature of the extracellular matrix proteins is also important in the function and development of the islet.37,38 Regional heterogeneity is seen in the pattern of endocrine cells in islets, for example, with glucagon-secreting α cells more abundant in the body and the tail of the pancreas, in contrast to the more frequent PP-secreting cells present in the head of the pancreas.39,40 Islets are richly vascularized, receiving ≈10% of pancreatic blood flow, despite accounting for only ≈1% of pancreatic mass.27,41–43 Islets are also richly innervated by nerve fibers tracking with vessels44–46 (Fig. 33-4). The arteriolar input to the islet initially supplies the β cell–rich islet core before it is further distributed to the α cell and/or the PP-enriched mantle.42 The consequence of this is that non–β cell endocrine cells in the islet are exposed to very high paracrine insulin concentrations that may be important in normal function.47–49 The development of the endocrine pancreas is addressed in detail elsewhere (see Chapter 31).50 It is clear that β cell mass is regulated in adult rodents, increasing in response to hyperglycemia, obesity, and pregnancy.51–54 β-Cell mass is also greater in obese versus lean humans, but the increment is much less marked (≈0.5-fold vs. ≈10-fold) than in obese rodents.55 Although an adaptive increase in β cell mass in mice is largely accomplished by an increase in β cell replication, β cell replication in vivo is very rare in humans.51,54,55 Because β cell replication appears to be rare in adult humans in contrast to mice, the question arises as to whether there is an alternative source of new β cells in adult humans. It has been proposed that new islets may be formed during adult life from ductal precursors recapitulating the pattern observed during development.50,56,57 Although islet buds are frequently seen on exocrine ducts in adult life in humans and rodents,55,57 it is difficult to prove that these are newly forming islets rather than products of arrested development. Interest now is focused on the possibility that stem cells might provide an ongoing source of β cells in health and may be harnessed therapeutically in diabetes. Data are conflicting regarding whether marrow-derived stem cells are potential precursors for β cells in rodents.58,59 As yet, no data are available on humans.
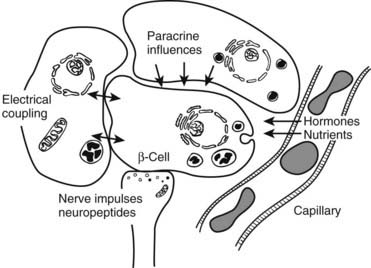
FIGURE 33-3. Schematic diagram of β cell with vascular, neural, and paracrine influences.
(Adapted with permission from Hellerstrom C: The life story of the pancreatic β cell. Diabetologia 26:395, 1984.)
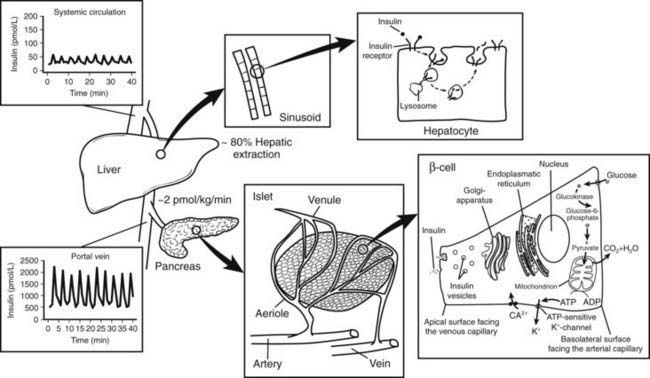
FIGURE 33-4. Relationship between insulin secretion from the islet, insulin clearance, and insulin action at the hepatocytes. Insulin is secreted at a rate of approximately 2 pmol/L/min from the islets of Langerhans into the portal vein. Approximately 80% of the total amount of insulin secreted is extracted from the liver sinusoids. Thus, oscillations in insulin secretion in the portal vein (≈2000 pmol/L) largely exceed those measured in the peripheral circulation (≈50 pmol/L). Following insulin binding, the insulin receptor-ligand complex is internalized into the cytosol of the hepatocyte. While insulin mainly undergoes enzymatic degradation in the lysosomes, the insulin receptor is reinserted into the plasma membrane within approximately 5 minutes.
Islet Function in Health
Given the critical importance of avoiding hypoglycemia, it is not surprising that the circulating glucose concentration is so predominant in the regulating of insulin secretion. Indeed the glucose dose response curve for insulin secretion by isolated human islets is remarkably similar to that of humans in vivo.60–62 So that beta cells can “sense” the prevailing blood glucose, islets need to be well vascularized, and the cytosol of β cells should be readily accessible to glucose. This is accomplished by rich islet vascularization with fenestrated vessels and abundant glucose-2-transporter proteins on the β cell surface.27,41–43,63,64 The latter allow rapid equilibrium of glucose between extracellular and intracellular concentrations. Given this rapid access of circulating glucose to the β cell cytosol, β cells “sense” the circulating glucose concentration by the rate-limiting step in glucose metabolism—phosphorylation of glucose to glucose-6-phopshate.65,66 This is accomplished in β cells by the expression of a glucokinase isoform with a Km of ≈150 mg/dL (7 mM) in the middle of the physiologic glucose concentration range.67 Thus the rate of provision of glucose-6-phopshate into the glycolytic pathway and the subsequent provision of pyruvate for the tricarboxylic acid cycle are closely linked to the plasma glucose concentration.66 The resultant mitochondrial pyruvate oxidation generates adenosine triphosphate (ATP), which in turn activates ATP-sensitive potassium channels (closing these channels), leading to cell depolarization and an influx of ionized calcium.68,69 The ionized calcium is believed to interact with primed docked insulin secretory vesicles that then discharge their contents either wholly (by exocytosis) or in part (by kiss and run) into the extracellular space.70,71 This rich vascular supply and the fenestrated vessels ensure rapid delivery of secreted insulin into the pancreatic venous efflux and then to the hepatic portal vein.
When islets are stimulated by an abrupt increase in glucose concentration in vitro (perifusion) or in vivo (intravenous glucose tolerance test), the resultant insulin secretion is biphasic60,72–74 (Fig. 33-5). An immediate first phase of insulin secretion occurs over ≈3 minutes and is followed by a more prolonged second phase of insulin secretion. This observation led to the concept proposed by Grodsky of distinct subcellular pools of insulin.5,75,76 More recently, these hypothetical pools have developed a likely anatomic basis. First-phase insulin secretion appears to reflect the immediate discharge of primed and docked insulin secretory vesicles, while second-phase insulin secretion most likely requires priming and mobilization of insulin vesicles before the time of their discharge.77 The exact molecular processes involved in the priming and mobilization of insulin vesicles remain unknown but may include provision of ATP following mitochondrial oxidation of pyruvate. An ATP-independent pathway for glucose-mediated insulin secretion also has been proposed, given the observation that when the KATP channel is defective as the result of mutations in the sulphonylurea receptor, some degree of glucose-mediated insulin secretion prevails.78
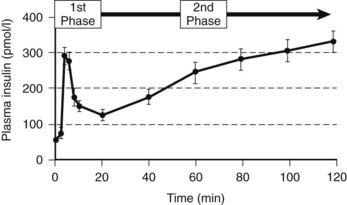
FIGURE 33-5. The relationship between the timing of the introduction of an intravenous glucose infusion (t = 0 min) and plasma insulin concentrations. A rapid first phase of insulin secretion is followed by second-phase secretion.
(From Pratley RE, Weyer C: The role of impaired early insulin secretion in the pathogenesis of type II diabetes mellitus. Diabetologia 44:931, 2001.)
It has been argued that there is no physiologic counterpart of first-phase insulin secretion in vivo, given the intravenous glucose challenge used to elicit it. To the contrary, almost all insulin secretion in vivo is likely released from the same pool, as ≈90% of insulin secretion is derived from discrete insulin secretory bursts that occur at ≈4 minute intervals.79,80 Thus regulation of ≈90% of insulin secretion can be accomplished through changes in size (secretory burst mass) or frequency of these discrete insulin pulses. The pacemaker for this high-frequency pulsatile insulin secretion is unknown, although it is present in individual islets, in that isolated independent islets secrete insulin in pulses every ≈4 minutes.60,73,81,82 Whatever the basis for the pacemaker, it is remarkably robust and does not appear to change under almost any conditions. Under almost all conditions studied, regulated changes in insulin secretion are accomplished exclusively through changes in the insulin secretory burst mass. For example, enhanced insulin secretion as a result of glucose ingestion or glucose infusion, infusion or ingestion of sulphonylurea drugs, and GLP-1 infusion is accomplished by an increase in the insulin secretory burst mass60,83–86 (Fig. 33-6). Suppression of insulin secretion by somatostatin and insulin-like growth factor-1 (IGF-1) is accomplished by a reduction in insulin burst mass.84,87 The one circumstance in which pulse frequency has been shown to change is induction of general anesthesia. Induction of general anesthesia profoundly suppresses insulin secretion, but although this is accomplished by inhibition of insulin burst mass, insulin pulse frequency increases under these circumstances.88
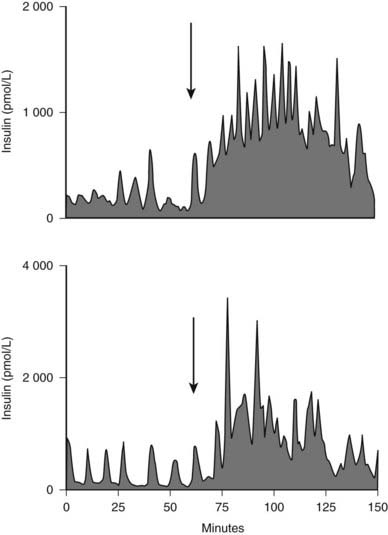
FIGURE 33-6. The portal vein insulin concentration before (0-60) and after meal ingestion (arrow) in two representative dogs. The portal vein insulin concentration excursions vary from approximately 300 to 1000 pmol/L before meal ingestion to 2000 to 4000 pmol/L after meal ingestion. Similar concentration profiles have been seen in the human portal vein.
(With permission from Porksen N, Munn S, Steers J, Veldhuis JD, Butler PC: Effects of glucose ingestion versus infusion on pulsatile insulin secretion: The incretin effect is achieved by amplification of insulin secretory burst mass. Diabetes 45:1317–1323, 1996.)
Because most insulin secretion in vivo arises from these discrete insulin secretory bursts, the approximately one million islets scattered in the exocrine pancreas must be coordinated to discharge their insulin secretory bursts synchronously. This coordination is accomplished at least in part by the intrinsic neural network in the pancreas, analogous to the intrinsic neural network in the gut, which allows coordinated peristalsis; it probably also occurs through entrainment by the oscillating glucose concentration, which presumably arises as a consequence of insulin pulses.89–93 As a consequence of this coordination, the insulin concentration wavefront that affects the liver each ≈4 minutes measures ≈2000 pmol/L in the fasting state and as much as 5000 pmol/L after meal ingestion.94 The amplitude of this concentration wavefront is greatly attenuated (≈50 pmol/L) by the time the insulin is released into the systemic circulation, presumably as a result of both dilution of portal vein insulin in the systemic circulation and selective extraction of insulin pulses in the liver (see Figs. 33-4 and 33-6). Although no studies have knowingly reproduced these dramatic portal vein insulin concentration dynamics in vivo, early-phase insulin secretion after glucose ingestion, which likely approximates these kinetics in the portal vein, may be important in suppressing hepatic glucose production, an important adaptive response to ingested glucose to minimize the postprandial increase in glucose concentrations (see Chapter 36). It is unknown to what extent exposure of the liver to these dramatic oscillations in insulin concentration is important for insulin sensitivity. Infusions of much smaller insulin pulses versus a continuous insulin infusion have been shown to enhance insulin action.93,95,96 Insulin is also secreted in an ultradian rhythm with a frequency of ≈20 minutes.97
Under conditions of daily living, numerous factors involved in regulating insulin secretion are integrated to provide a rate of insulin secretion of ≈2 pmol/kg/min in the fasting state, increasing to ≈10 pmol/kg/min after meal ingestion.98–101 The wide range in these rates is based on the insulin sensitivity of the individual (see Chapter 41). Thus with aging and obesity, and in response to exercise, adaptive changes in the rate of insulin secretion occur.79,101–107 In health, insulin secretion adaptively changes according to insulin requirements.62,104,108 The most prevalent need for increased insulin secretion involves the insulin resistance consequent upon obesity.79,101 In response to obesity, the daily insulin requirement increases by as much as 10-fold. However, in humans, the β cell mass in obese versus lean individuals is increased only ≈0.5-fold.55 This implies that the most important adaptive change needed to meet chronically increased insulin secretion requirements in insulin-resistant humans is an increase in insulin secretion per β cell, rather than simply an increase in the number of β cells.
Insulin Clearance
Insulin is secreted in the portal vein, where it is delivered directly to the liver. As a consequence of rapid blood flow in the portal vein (≈0.8 L/min) and fenestrations in the hepatic sinusoids, hepatocytes are directly exposed to the high-amplitude oscillations that arise from insulin secretory bursts within seconds of secretion. After insulin binding to the receptors at the hepatocyte membrane, the insulin receptor-ligand complex is rapidly internalized to form an intracytoplasmic vesicle.109–114 Although insulin undergoes enzymatic degradation in the endosomes,115,116 the insulin receptor is reinserted into the plasma membrane within ≈5 minutes to become available for the next insulin burst.113,117 In health in humans, ≈80% of endogenous insulin secretion is cleared by the liver with the first pass through the liver.118–121 Following oral glucose ingestion, insulin extraction is diminished by ≈50%.122–124 The major factor that determines the rate of insulin clearance appears to be the amount of insulin presented to the liver.125,126 In fact, a close relationship between insulin secretion and hepatic insulin uptake has been described,119,127 consistent with the idea of a finite number of insulin receptors present on hepatocytes. The extent of hepatic extraction also appears to depend on the amplitude of insulin oscillations presented to the liver.84,128 Since the mean interval of insulin pulses presented to the liver almost coincides with the time period calculated for insulin receptor recycling (≈5 minutes),113 pulsatile delivery of insulin may prevent the liver from desensitization. This, together with the fact that selective extraction of insulin pulses by the liver is evident, implies that varying the pattern of insulin delivery to the liver may provide the β cell an opportunity to regulate end-organ actions of insulin.
Islet Structure and Function in Type 1 Diabetes
In type 1 diabetes, a marked deficiency in β cell mass is seen.56,129–133 Before diabetes develops, a prolonged period occurs during which the autoimmune disease is thought to be active.134–136 During this latent period, a progressive decline in first-phase insulin secretion has been documented, as have impaired insulin pulses.135,137–140 By the time of clinical presentation, ≈90% of β cells have been lost; however, a substantial capacity for endogenous insulin secretion remains, although a relatively rapid further loss of this occurs over the next 2 years.56,131,133,141,142 The mechanisms underlying this further loss of insulin secretion likely include both further loss of β cell mass and decreased insulin secretion per β cell, each in large part a consequence of the hyperglycemia.143,144 It is important here to distinguish human islets from rodent islets, about which of necessity much research is carried out. Human β cells exposed to glucose concentrations typically present in patients with even relatively well-controlled diabetes (≈150 mg/dL; 8 mM) have an increased frequency of apoptosis, induced in part by endogenous expression of interleukin-1β (rat islets do not have an increased rate of β cell apoptosis until glucose concentrations increase to ≈360 mg/dL or 20 mM).143 In addition, insulin secretion by human islets is impaired within 96 hours after exposure to these levels of glucose concentration.145 Chronic exposure of islets to this relatively modest level of glucose appears to preferentially deplete the primed and docked insulin secretory vesicles in that both glucose-induced first-phase insulin secretion and glucose-induced insulin secretory burst mass are greatly attenuated.145 Evidence also suggests that an increased workload of β cells may accelerate the autoimmune-mediated destruction. Taken together, these factors have been called glucose toxicity.144,146 The reversibility of these factors at least in the short term is illustrated by the partial recovery of insulin secretion and glycemic control in patients treated who are brought toward normal blood glucose concentrations with exogenous insulin therapy.147–150
Not only is some residual insulin secretion detected in patients with recent-onset type 1 diabetes, but with increasingly sensitive assays, insulin secretion may be detected many years after onset of type 1 diabetes. Moreover, detectable β cells are commonly present in the pancreas of patients with even longstanding type 1 diabetes.56 The question arises whether this residual insulin secretion results from a small pool of β cells that are relatively protected from β cell destruction, or from newly formed β cells. This distinction is important because the latter would imply that a novel approach to restoration of β cell mass in patients with type 1 diabetes would be to suppress ongoing β cell destruction. Preservation or restoration of even an inadequate amount of insulin secretion to allow insulin independence still would have great potential clinical benefit in that microvascular complications are decreased in patients with residual β cell function.151
Therefore to summarize, it is clear that impaired insulin secretion in patients with type 1 diabetes is as a consequence of a major defect in β cell mass, but functional defects likely arise from the ongoing inflammation and glucose toxicity once diabetes supervenes.
Islet Structure and Function in Type 2 Diabetes
Most but not all studies indicate that there is an ≈60% decrease in β cell mass in humans with type 2 diabetes (Fig. 33-7).55,152,153 This decrease in β cell mass appears to be due to an increased frequency of β cell apoptosis; therefore type 2 diabetes can be considered to have much in common with type 1 diabetes.54,55,154,155 The most important distinction appears to be the absence of an autoimmune-mediated cause for the accelerated β cell apoptosis and the more modest degree of β cell deficiency. The importance of an ≈60% deficit in β cell mass might be questioned because a similar defect does not lead to diabetes in rodents.156 Although juvenile rodents have a remarkable capacity for β cell regeneration after a partial pancreatectomy,51,54 surgical reduction of β cell mass in adult humans does not prompt β cell regeneration.157 A deficit in β cell mass comparable with that seen in humans with type 2 diabetes does lead to diabetes in large animal species that are potentially more representative of humans, including the pig, the dog, and nonhuman primates.128,158–160 Indeed a comparable β cell deficit leads to loss of first-phase insulin secretion, a deficit in insulin pulse mass, and decreased hepatic insulin clearance in the pig,128,161 thereby reproducing the pattern of abnormal insulin secretion and insulin clearance seen in type 2 diabetes.162 The increased frequency of β cell apoptosis in type 2 diabetes has been ascribed to glucose toxicity and to increased concentrations of free fatty acids, free radicles, and oligomers of islet amyloid polypeptide (IAPP, also known as amylin).143,163,164 The key question as to whether this increase precedes the development of hyperglycemia or is a consequence of it remains unknown.
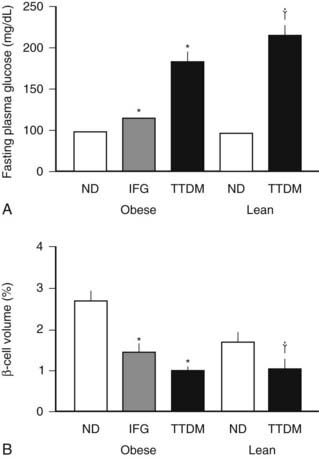
FIGURE 33-7. The mean blood glucose (A) and relative pancreatic β cell volume (B) in obese versus lean human subjects with type 2 diabetes (TTDM), those with impaired fasting glucose (IFG), and nondiabetic patients (ND). The relative β cell volume is increased in obese versus lean ND by approximately 50%. The relative β cell volume is decreased by approximately 65% in obese TTDM versus controls.
(From Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC: β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52:105, 2003.)
The functional defects in insulin secretion seen in type 2 diabetes have been reviewed elsewhere.74 In brief, when glucose concentrations are matched, there is a major defect in insulin secretion in both basal and glucose-stimulated (hyperglycemic clamp or oral glucose load) insulin secretion165–169 (Figs. 33-8 through 33-10). Defects in insulin secretion in response to different stimuli, including glucose, arginine, and GIP, can be detected in individuals at high risk for developing type 2 diabetes, such as first-degree relatives or women with a history of gestational diabetes.166,170–176 A decrease in pancreatic insulin stores is noted in patients with type 2 diabetes, but given the small proportion of insulin vesicles that undergo secretion, it is clear that there must be a specific defect in the availability of primed, docked insulin secretory vesicles for glucose stimulation to occur.77 This is supported by marked defects in early insulin secretion after meal ingestion, first-phase insulin secretion after glucose ingestion, and defective glucose-mediated insulin secretory burst mass in type 2 diabetes (see Figs. 33-10 and 33-11).74,177–179 The increased ratio of circulating proinsulin to insulin characteristic of type 2 diabetes has been ascribed to both defective proinsulin processing and increased insulin demand leading to secretion of immature insulin vesicles.180–182 The defects in first-phase insulin secretion, insulin pulse mass, and proinsulin/insulin processing all can be reversed in patients with type 2 diabetes by overnight inhibition of insulin secretion (see Fig. 33-11).183 The pattern of insulin secretion defects present in type 2 diabetes can be recapitulated in pigs through induction of a deficit in β cell mass comparable with that seen in type 2 diabetes.128,161 It is of interest to compare the normal adaptive responses of β cell mass and insulin secretion to insulin resistance in obese nondiabetic humans versus the deficits in these parameters in obese humans with type 2 diabetes (Fig. 33-12). In nondiabetic humans, a modest adaptation in β cell mass (≈50% increased) and a much greater increase (≈300%) in insulin secretion are seen, so that in the setting of an adequate β cell mass and normal blood glucose concentrations, β cells show considerable capacity for sustained increased secretion. In contrast, in type 2 diabetes, a rather comparable deficit in β cell mass and insulin secretion (≈60%) is noted under conditions of daily living (as in Fig. 33-12), although the deficit in insulin secretion can be considered much greater at matched glucose concentrations (as in Fig. 33-8).
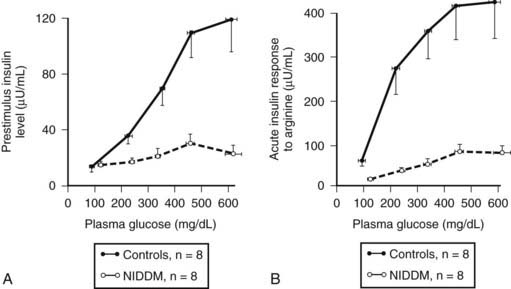
FIGURE 33-8. The plasma insulin concentration in patients with type 2 diabetes (NIDDM) and nondiabetic controls in relation to a graded glucose infusion (A) and following an arginine bolus at graded glucose concentrations (B), revealing marked impairment of insulin secretion to both glucose and arginine when glucose values are matched.
(With permission from Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr: Diminished β cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 74:1318, 1984.)
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


