FIGURE 13-1. Standard mortality rates (SMR) and 95% confidence intervals (CI) in individual studies on patients with nonmalignant pituitary diseases not associated with excess adrenocorticotropic hormone (ACTH) or growth hormone (GH) secretion, and in the weighted meta-analysis (bottom line). Results are shown for men (open boxes) and women (black boxes) separately.
(From Nielsen EH, Lindholm J, Laurberg P. Excess mortality in women with pituitary disease: a meta-analysis. Clin Endocrinol 67:693–697, 2007.)
GH deficiency has been implicated as a major contributor to excess mortality in hypopituitarism because it is the only defect not replaced in the studies of hypopituitarism. However, the contribution to overall mortality of other risk factors, such as radiotherapy and suboptimal replacement therapies for other hormone deficits, is the subject of ongoing investigation.
Causes
Major causes of hypopituitarism are shown in Table 13-1. The most common cause is a pituitary adenoma or treatment with pituitary surgery or radiotherapy.
Table 13-1. Causes of Hypopituitarism
PITUITARY AND HYPOTHALAMIC MASS LESIONS
Pituitary adenomas account for the vast majority of pituitary mass lesions, although secondary tumors do occur, from metastases to the pituitary gland from carcinomas of the breast, lung, colon, and prostate. Pituitary microadenomas are surprisingly common and are found in between 1.5% and 27% of patients at autopsy15; these tumors are very rarely, if at all, associated with hypopituitarism and tend to run a benign course. Macroadenomas are less common but are more frequently associated with pituitary hormone deficiencies; some 30% of patients with pituitary macroadenomas have one or more anterior pituitary hormone deficiencies. The causative mechanism of hypopituitarism is compression of the portal vessels in the pituitary stalk, secondary to the expanding tumor mass directly or to increased intrasellar pressure,16 which explains the potential reversibility of pituitary dysfunction after surgery in some patients.
Many hypothalamic mass lesions arise from developmental abnormalities such as craniopharyngiomas, Rathke’s cleft cysts, and arachnoid cysts. Craniopharyngiomas, the third most common intracranial tumor, account for most parapituitary tumors that occur. A bimodal peak in incidence occurs at 5 to 14 years and again after the age of 50. Their development origin is uncertain. They frequently have large cystic components and may be intrasellar, extrasellar, or both. Rathke’s cleft cysts are cystic sellar and suprasellar lesions lined by a single epithelial layer. Arachnoid cysts present at a later age and are less common than both craniopharyngiomas and Rathke’s cleft cysts.
Derangement of central endocrine regulation also occurs with other parapituitary space-occupying lesions such as chondromas, chordomas, suprasellar meningiomas, astrocytomas of the optic nerve, and primary tumors of the third ventricle.
Pituitary Surgery
The incidence and degree of hypopituitarism after surgery depend on the size of the original tumor, the degree of infiltration, and the experience of the surgeon. About 50% of patients already had evidence of GH, gonadotropin, or cortisol deficiency17 before surgery. The patient should also be warned of a possible deterioration of postoperative pituitary function, and assessment of pituitary function should be performed promptly after surgery. However, a postoperative decline in pituitary function is not universal. After surgery, about 80% had evidence of GH or gonadotropin deficiency. In patients who received postoperative radiotherapy, evaluation after 5 years revealed that all patients were GH deficient.17 On the other hand, surgery for pituitary adenomas may be associated with significant recovery of pituitary function. About half of the patients recover at least one pituitary insufficiency after transsphenoidal surgery. Postoperative improvement is more likely if no tumor is found on postoperative imaging, or if the tumor is not invasive.18 The pituitary hormone most likely to recover is thyroid-stimulating hormone (TSH), followed in order by adrenocorticotropic hormone (ACTH), gonadotropins, and GH.19 Recovery of pituitary function occurs early, within 8 weeks after surgery.20
Radiotherapy
Deficiency of one or more anterior pituitary hormones is almost invariable when the hypothalamic-pituitary axis lies within the fields of radiation. Hypopituitarism also develops in patients who received radiation therapy for nasopharyngeal carcinomas, parasellar tumors, and primary brain tumors, as well as in children who underwent prophylactic cranial irradiation for acute lymphoblastic leukemia or total body irradiation (TBI) for a variety of tumors and other diseases.21
The radiobiological impact of an irradiation schedule is dependent on the total dose, the number of fractions, and the duration and length of follow-up. Somatotrophs are the most sensitive to radiation damage, and thus, after lower radiation doses (<30 Gy), isolated GH deficiency ensues, whereas higher doses (30 to 50 Gy) increase the frequency of GH deficiency to 50% to 100% and may produce panhypopituitarism (Fig. 13-2). Radiation dose also determines the speed of onset of hormonal deficiency. The greater the dose, the earlier is the occurrence of GH deficiency, so that between 2 and 5 years after irradiation, 100% of children receiving more than 30 Gy (over a 3-week period) to the hypothalamic-pituitary axis developed subnormal GH responses to an insulin tolerance test (ITT), whereas 35% of those receiving less than 30 Gy (over a 3-week period) still showed a normal GH response22 (Fig. 13-3). However, interpretation of the impact of radiation-induced damage to the hypothalamic-pituitary axis on GH status is complicated in the early years after irradiation. Discordant results to different GH-provocative agents may be seen, such that up to 50% of patients classified as severely GH deficient with use of the ITT showed normal or mildly insufficient GH response during the combined GH-releasing hormone (GHRH) plus arginine stimulation test.23,24 The discordant response to dynamic testing in patients with GH deficiency is discussed in greater detail later in the chapter, under Diagnosis in Growth Hormone Deficiency.
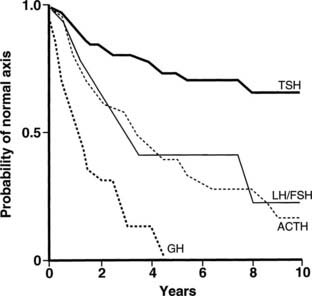
FIGURE 13-2. Life-table analysis indicating probabilities of initially normal hypothalamic-pituitary-target gland axes remaining normal after radiotherapy (3750 to 4250 cGy). Growth hormone (GH) secretion is the most sensitive of the anterior pituitary hormones to the effects of external radiotherapy, and thyroid-stimulating hormone (TSH) secretion is the most resistant. In two thirds of patients, gonadotropin deficiency develops before adrenocorticotropic hormone (ACTH) deficiency. FSH, Follicle-stimulating hormone; LH, luteinizing hormone.
(From Littley MD, Shalet SM, Beardwell CG, et al: Hypopituitarism following external radiotherapy for pituitary tumors in adults. Q J Med 70:145–160, 1989.)
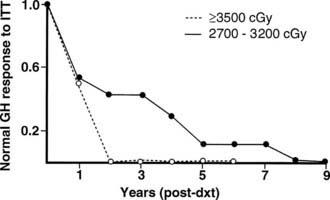
FIGURE 13-3. The incidence of growth hormone (GH) deficiency in children receiving 27 to 32 Gy or ≥35 Gy of cranial irradiation for a brain tumor in relation to time from irradiation (dxt). This illustrates that the speed at which individual pituitary hormone deficits develop is dose dependent; the higher the radiation dose, the earlier GH deficiency occurs.
(Courtesy the Department of Medical Illustrations, Withington Hospital, Manchester, England.)
Paradoxically, whereas high doses of cranial irradiation may render a child gonadotropin-deficient, lesser doses of irradiation may be associated with early puberty. The mechanism for early puberty after irradiation is likely to be related to disinhibition of cortical influences on the hypothalamus.
Fractionated stereotactic conformal radiotherapy (SCRT) is a more precise technique of localized irradiation that may reduce radiation damage to normal structures in the brain. However, despite this theoretical advantage of normal-tissue sparing, hypopituitarism remains a common complication. At a median follow-up of 32 months, 22% of patients with previously normal pituitary function or partial hypopituitarism developed new hormonal deficit, and 18% developed panhypopituitarism.25 The incidence of hypopituitarism was not different between conventional radiotherapy and SCRT,25 and it may occur as late as 10 years after therapy, reaching as high as 66% after a median follow-up of 17 years.26,27 Therefore, with increased survival, follow-up evaluation of patients irradiated for tumors of the brain and surrounding structures must focus equally on the possibility of tumor recurrence and on the delayed effects of therapy, including the endocrine effects. Endocrine testing should be performed on a yearly basis for at least 10 years and again at 15 years.
GENETIC CAUSES
Major advances in our understanding of the developmental biology of the pituitary in recent years have provided insights into the molecular pathology of genetic causes of hypopituitarism. During embryonic development, the pituitary gland is formed by the association of neural ectodermal cells from the ventral diencephalons with ectodermal cells from the oral cavity. The former give rise to the posterior pituitary, and the latter give rise to the anterior pituitary. The formation and differentiation of Rathke’s pouch, the primordial anterior pituitary lobe structure, into the mature pituitary gland are regulated by the actions of specific transcription factors in a spatial and temporal fashion. These include KAL, HESX-1, Prop-1, and Pit-1. Mutations in genes encoding for these transcription factors result in deficiency of one or more anterior pituitary hormones.
Combined Pituitary Hormone Deficit
A cascade of pituitary transcription factors regulates the differentiation of cells of Rathke’s pouch into somatotrophs, lactotrophs, thyrotrophs, gonadotrophs, and corticotrophs. Mutations in early appearing transcription factors tend to cause more extensive hormone deficiencies (Table 13-2).
Table 13-2. Genetic Causes of Hypopituitarism
| Gene Defect | Hormone Deficiencies | |
|---|---|---|
| Combined | Pit-1 (POU1F1, GHF1) | GH, TSH, PRL |
| PROP-1 | GH, LH/FSH, TSH, ACTH, PRL | |
| HESX1 (Rpx) | GH, LH/FSH, TSH, ACTH, ADH | |
| LHX3/LHX4 | GH, LH/FSH, TSH, PRL | |
| PITX2 | GH, PRL | |
| Isolated | hGH | GH |
| GHRH receptor gene | GH | |
| KAL | FSH/LH | |
| GnRH receptor gene | FSH/LH | |
| DAX1/AHC | FSH/LH | |
| TBX19 (Tpit) | ACTH | |
| TSH-β gene | TSH | |
| TRH receptor gene | TSH |
Pit-1 (pituitary-specific transcription factor-1) is a pituitary-specific transcription factor that is responsible for the development of somatotrophs, lactotrophs, and thyrotrophs in mammals. Pit-1 abnormalities account for only a small minority of the total number of worldwide cases of hypopituitarism. Pit-1 gene mutations also have been discovered in patients with idiopathic GH deficiency associated with preserved basal prolactin and TSH secretion, illustrating the variability of phenotypic presentation among these patients.
PROP1 (Prophet of Pit-1) is a novel pituitary paired-like homeodomain factor, which regulates the expression of Pit-1. Several multicenter studies of patients have reported that PROP1 mutations are the most common genetic cause of multiple pituitary hormone deficiencies, accounting for up to 40% to 50% of cases. PROP1 abnormalities result in similar phenotypic abnormalities to Pit-1 mutations but with associated gonadotropin deficiency. The degree of gonadotropin deficiency is variable, even among individuals within the same family with identical mutations. In individuals with a mutation of the PROP1 gene, progressive hypopituitarism develops, with GH, TSH, and gonadotropin deficiency typically present by the end of the second decade.28 The pituitary gland may pass through a hyperplastic phase before undergoing a phase of degeneration and late appearance of partial ACTH deficiency.
Other genetic causes of combined pituitary hormone deficiencies include mutations of HESX1, LHX3/LHX4, and Pitx1/Pitx2, which are transcription factors engaged in early pituitary cell development before the appearance of Pit-1 and PROP1.29–31 For example, HESX-1 is a homeobox gene expressed early in the ectoderm, which is the precursor of Rathke’s pouch. It plays an important role in optic nerve and anterior pituitary development. HESX-1 mutations in humans are associated with septo-optic dysplasia and GH deficiency. Other genes (e.g., LHX3) bind to Pit-1 and can enhance Pit-1 activity, or synergistically activate prolactin and TSH genes (e.g., Pitx1).
Isolated Pituitary Hormone Deficiency
Isolated Growth Hormone Deficiency
Isolated GH deficiency (IGHD) can arise from mutations of the GH gene and of the GH-releasing hormone (GHRH) receptor gene.32 The human GH (hGH) gene is located on chromosome 17 in a cluster of five genes: hGH-N encodes the gene for pituitary GH, hGH-V encodes the gene for placental GH, and three genes encode for human chorionic somatotropin (hCS). There are four types of Mendelian disorders of the GH gene that are due to deletion of these genes: IGHD IA and IB are both inherited in an autosomal recessive manner, resulting in absent or low GH levels. In patients with absent GH (IGHD IA), anti-GH antibodies often develop when they are treated with GH. IGHD II has an autosomal dominant mode of inheritance with variable clinical severity. IGHD III is an X-linked disorder that often is associated with hypogammaglobulinemia. Mutations of the gene encoding the GHRH receptor have been identified in a number of kindreds with severe GH deficiency in Pakistan and in Brazil.33,34
Isolated Gonadotropin Deficiency
Several gene mutations have been identified as causes of idiopathic hypogonadotropic hypogonadism (IHH) in humans.35 Gonadotropin-releasing hormone (GnRH) neurons originate in the olfactory placode and migrate during embryogenesis with the olfactory nerves to the hypothalamus. The KAL protein is necessary for this process to occur. Kallmann’s syndrome is characterized by the combination of IHH and anosmia or hyposmia, which usually is caused by defective GHRH secretion. It is a heterogeneous condition that manifests in an X chromosome–linked or autosomal dominant form. The X-linked form of the disorder is due to mutations in the KAL gene. Recently, several growth factors, including fibroblast growth factors (FGFs) and adhesion molecules, have been identified to play important regulatory roles in the development and migration of GHRH neurons. Mutation of the FGF receptor 1 (FGFR1) gene causes the autosomal dominant form of Kallmann’s syndrome.36 Additional clinical features associated with KAL mutations include bimanual synkinesia and renal agenesis; FGFR1 mutations typically lead to cleft lip palate and dental agenesis.
Another important protein is DAX1, which is a transcription factor involved in development of pituitary gonadotrophs and the adrenal cortex. Mutations in the DAX1 gene give rise to X-linked recessive hypogonadotropic hypogonadism and adrenal hypoplasia. Other genes implicated in IHH are PC1 (prohormone convertase, associated with defects in prohormone processing), OB, and DB (leptin and leptin receptor, associated with obesity), whereas inactivating mutations of luteinizing hormone (LH)-β and follicle-stimulating hormone (FSH)-β genes can cause isolated deficiencies of LH and FSH, respectively.
Isolated ACTH and TSH Deficiencies
Isolated deficiencies of TSH or ACTH are very rare; however, in a number of cases, a genetic abnormality has been described or proposed. Mutations of the coding region of the TSH-β subunit gene37 and the thyrotropin-releasing hormone (TRH)-receptor gene38 have been found in a number of families to be the cause of hereditary isolated TSH deficiency.
Recently, a pituitary transcription factor causing isolated ACTH deficiency was identified. TBX19 (the human T-box pituitary transcription factor, analogous to Tpit in the mouse) plays an essential role in differentiation of pro-opiomelanocortin (POMC) cells in the pituitary. At least two TBX19 gene mutations causing isolated ACTH deficiency have been described,39,40 and this may underlie the cause of a neonatal-onset form of congenital isolated ACTH deficiency,41 which is fatal unless diagnosed early and replaced with glucocorticoid.
Traumatic Brain Injury
Traumatic brain injury (TBI) is an under-appreciated cause of hypopituitarism. It was first reported in 1918. Meta-analysis of 19 studies, which included more than 1000 patients, demonstrated a pooled prevalence of hypopituitarism following TBI of 27.5% (1 pituitary hormone deficiency), and 7.7% of patients had multiple deficiencies. GH deficiency was the most common with a prevalence of 12.4%, followed by secondary hypogonadism (12.5%), hypoadrenalism (8.2%), and hypothyroidism (4.1%). Prevalence of diabetes insipidus is 26% in the acute phase and is decreased to 6.9% among long-term survivors.42
Risk factors of traumatic hypopituitarism include basal skull fracture, diffuse axonal injury, raised intracranial pressure, and prolonged stay in the intensive care unit.
TBI is common, with an overall incidence of 235 per 100,000 persons per year.43 Traumatic hypopituitarism therefore is a major public health issue with significant clinical implications. The incidence of hypopituitarism following TBI is more than 30 patients per 100,000 population per year.44 Patients therefore should be screened for hypopituitarism following TBI. Early (<3 months) hormonal dysfunction, including central hypothyroidism and hypogonadism, was not predictive of long-term development of hypopituitarism.45 Assessment for GH deficiency, hypogonadism, and hypothyroidism is not necessary in the acute phase, but adrenal insufficiency should not be missed, as it can be fatal if untreated. All patients should undergo screening for hypopituitarism between 3 and 6 months after injury.
Lymphocytic Hypophysitis
Lymphocytic hypophysitis, an immune-mediated diffuse infiltration of the anterior pituitary with lymphocytes and plasma cells, occurs predominantly in women and often is first evident in pregnancy or after delivery. The classic presentation is peripartum hypopituitarism, often with a pituitary mass and visual failure. ACTH deficiency is an almost universal feature that, when undiagnosed, has proved fatal. At an early stage, the pituitary gland is enlarged and cannot be distinguished from a pituitary tumor by computed tomography (CT) or magnetic resonance imaging (MRI), whereas in later stages, the gland may atrophy, leaving an empty sella. Lymphocytic hypophysitis is more common in patients with other autoimmune endocrine diseases. Cytosolic autoantigens against the pituitary can be demonstrated in some cases but also are present in normal patients; thus the definitive diagnosis of this condition remains difficult without pituitary biopsy. Recently, identification of more specific autoantigens, such as chorionic somatomammotropin, may allow noninvasive diagnosis of hypophysitis in the future, with a sensitivity of 64% and a specificity of 86% by immunoblotting.46 Spontaneous resolution of both the mass and the hypopituitarism has been reported, and in some cases, neurosurgical intervention has led to irreversible pituitary failure. Therefore, conservative management is appropriate in most patients. Spontaneous recovery with physiologic hydrocortisone replacement can happen,47 and high-dose methylprednisolone pulse therapy may improve adrenopituitary function and shrinkage of the sellar mass.48
Pituitary Apoplexy
Pituitary apoplexy is the abrupt destruction of pituitary tissue that results from infarction or hemorrhage into the pituitary, usually into an underlying pituitary tumor. Severe headache accompanies a variable degree of visual loss or cranial nerve palsy. Consequent pituitary hormone deficiencies may develop rapidly. In Sheehan’s syndrome, pituitary infarction occurs secondary to severe postpartum hemorrhage and ensuing circulatory failure. Once common, this complication is now confined mainly to areas where obstetric services are less well developed.
Granulomatous Diseases
Granulomatous diseases, including sarcoidosis, tuberculosis, and Langerhans cell histiocytosis, can affect the hypothalamic-pituitary axis and cause hypopituitarism, including diabetes insipidus. Diabetes insipidus complicates sarcoidosis rarely (1%). This is more common, however, in Langerhans cell histiocytosis, with diabetes insipidus developing in 15% of childhood cases; it also may occur in patients first seen in adulthood.
Hypopituitarism
CLINICAL FEATURES
Presentation of hypopituitarism can be nonspecific. It is affected by degree, type, and rate of onset of the pituitary hormone deficiency. Local pressure effects or hormonal hypersecretion may complicate the clinical picture. Hypopituitarism arising from an expanding mass lesion or from irradiation produces a characteristic evolution of pituitary failure caused by an initial loss of GH secretion, followed by LH and FSH, and finally by failure of ACTH and TSH secretion.
In cases arising from loss of function caused by an expanding silent mass lesion, the onset of symptoms is insidious, typically occurring with mild headaches, lethargy, fatigue, disinterest, weight gain, low mood, and declining libido—symptoms mimicking depression. Rarely, anorexia and weight loss may arise from ACTH deficiency and may be mistaken for and lead to extensive investigations for occult malignancy. Progressive mass expansion causing increasingly severe headaches or visual symptoms from chiasmal compression usually leads to radiologic investigations that clinch the diagnosis. A high index of suspicion is required to diagnose hypopituitarism. The symptoms and signs of individual hormone deficiency are listed in Table 13-3. The features of isolated deficiencies of each axis are described below. GH deficiency is addressed separately in the section dedicated to GH deficiency in adults.
Table 13-3. Symptoms and Signs of Hormone Deficiencies
| Hormone Deficiency | Symptoms and Signs |
|---|---|
| Growth hormone | Please refer to Table 13-6 in the section Growth Hormone Deficiency. |
| Gonadotropins | In men: poor libido/impotence, infertility, small soft testes, reduced facial/body hair |
| In women: amenorrhea/oligomenorrhea dyspareunia, infertility, breast atrophy | |
| Thyroid-stimulating hormone | Growth retardation in children; decrease in energy; constipation; sensitivity to cold, dry skin, weight gain |
| Adrenocorticotropic hormone | Weakness, tiredness, dizziness on standing, pallor, hypoglycemia |
| Prolactin | Failure of lactation |
| Antidiuretic hormone | Polyuria, polydipsia, nocturia, hypotension |
Gonadotropin Deficiency
In male patients, the clinical features of gonadotropin deficiency differ according to whether the deficiency was acquired before or after pubertal age. If acquired before pubertal age, clinical examination reveals a small penis, small testes, and eunuchoid proportions (span exceeds a height of 5 cm). Hypogonadism acquired postpubertally is associated with a reduction in testicular size, loss of facial and body hair, and thinning of the skin, leading to the characteristic finely wrinkled facial skin of the “aging youth.” Other effects include a decrease in skeletal muscle mass, bone mineral density, sexual function, libido, and general well-being. Azoospermia is an almost inevitable consequence of hypogonadotropic hypogonadism. Partial LH deficiency may result in low circulating testosterone levels and gynecomastia with preserved testicular size and fertility, as intratesticular testosterone levels remain high enough to maintain spermatogenesis.
In a teenage girl, hypogonadotropic hypogonadism is associated with primary amenorrhea and absent breast development. In the adult woman, amenorrhea or oligomenorrhea, infertility, breast atrophy, vaginal dryness, and dyspareunia occur; pubic and axillary hair remains unless ACTH deficiency also is present.
Adrenocorticotropic Hormone Deficiency
ACTH deficiency is the most life-threatening component of hypopituitarism. If the onset is abrupt, as in pituitary apoplexy, the clinical picture may be dominated by profound shock in the most serious form. Patients with chronic ACTH deficiency usually present with chronic progressive symptoms of chronic fatigue, anorexia, and weight loss, sometimes mimicking anorexia nervosa or an occult malignancy. Patients on long-term glucocorticoid therapy can develop adrenal atrophy secondary to ACTH suppression.49,50 Examination may reveal pallor of the skin, in contrast to the hyperpigmentation of Addison’s disease, and in female patients particularly, loss of secondary sexual hair occurs. In severe ACTH deficiency, particularly in childhood, hypoglycemia can occur: Cortisol deficiency results in increased insulin sensitivity and a decrease in hepatic glycogen reserves. Hyponatremia, although less commonly seen than in Addison’s disease because of preservation of aldosterone secretion, may be the presenting feature of ACTH deficiency, particularly in the elderly.
Thyroid-Stimulating Hormone Deficiency
Thyroid-stimulating hormone (TSH) deficiency occurs late in most pituitary disorders. Symptoms include fatigue, weakness, inability to lose weight, constipation, and cold intolerance, in keeping with the symptoms of primary hypothyroidism. However, symptoms generally are milder than in primary hypothyroidism, because some residual TSH secretion often is preserved.
Antidiuretic Hormone Deficiency
Polydipsia and polyuria with nocturia are the classic features of diabetes insipidus resulting from antidiuretic hormone (ADH) deficiency. If the patient is unable to keep up with the fluid loss, hypotension and hypovolemia ensue. The features of diabetes insipidus may be masked by the presence of ACTH deficiency, because of the consequent hypovolemia and reduced glomerular filtration rate. Only when cortisol replacement therapy is commenced may the polyuria and polydipsia of diabetes insipidus be revealed.
DIAGNOSIS AND ENDOCRINE EVALUATION
Imaging
Computerized digital imaging has revolutionized the investigation of pituitary function, as it provides unparalleled views of the anatomy of the region, readily identifying structural abnormalities. MRI is the scanning technique of choice, as it offers higher resolution than CT scanning and is able to demonstrate microadenomas as small as 3 mm in diameter. MRI also has provided insights into the morphologic abnormalities that arise from developmental defects of the pituitary gland and how morphologic abnormalities relate to dynamic tests of pituitary function, especially in GH deficiency. CT is used in situations where MRI is contraindicated, such as when arterial clips or a pacemaker is present. CT has a valuable role in defining bone anatomy in preparation for surgery.
Radiologic Phenotype of GH Deficiency
GH deficiency can be divided broadly into either genetic GH deficiency, when proven genetic mutations, such as GH-1, GHRH-R, and PROP-1, are identified, or idiopathic GH deficiency, when a genetic abnormality cannot be identified. On MRI, cases of genetic GH deficiency typically reveal a pituitary gland that is small or of normal size. The pituitary stalk is intact, and the location of the posterior lobe is normal. In contrast, idiopathic GH deficiency frequently is associated with a small pituitary gland, with evidence of stalk hypoplasia or interruption and an ectopic posterior lobe (Fig. 13-4). It has been suggested that perinatal trauma may be responsible for these abnormalities on MRI, resulting in idiopathic GH deficiency.51,52 This is supported by the observed higher frequency of breech delivery and birth hypoxemia with idiopathic GH deficiency than in genetic cases.53
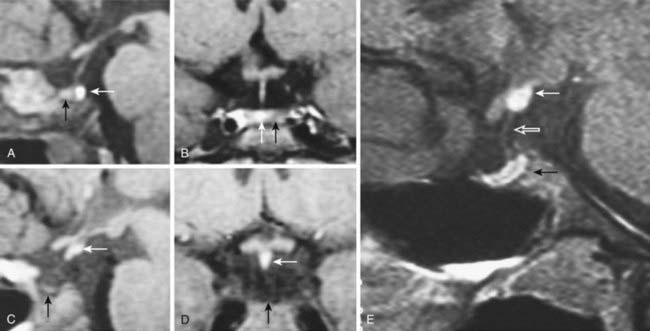
FIGURE 13-4. Magnetic resonance imaging (MRI) of congenital causes of growth hormone deficiency: Sagittal (A) and coronal (B) pituitary imaging studies of a patient with isolated growth hormone (GH) deficiency caused by GH-1 gene deletion of 6.7 kb showing intact stalk, normal pituitary gland (black arrow), and posterior lobe (white arrow); sagittal (C) and coronal (D) views of a patient with idiopathic isolated GH deficiency showing stalk interruption, an ectopic posterior lobe (white arrow), and pituitary gland hypoplasia (black arrow). Sagittal (E) view of pituitary MRI of a 17-year-old patient with idiopathic GH deficiency, demonstrating a hypoplastic gland (black arrow), stalk hypoplasia (hollow white arrow), and an ectopic posterior pituitary gland (white arrow).
(From Osorio MG, Marui S, Jorge AA, Latronico AC, Lo LS, Leite CC, Estefan V, Mendonca BB, Arnhold IJ: Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab 87:5076–5084, 2002.)
The stalk provides vascular communication, and its presence is significant in relation to the evaluation of diagnostic testing for GH deficiency. Maghnie and colleagues have reported that integrity of the hypothalamic-pituitary connection is necessary for GHRH-arginine to stimulate GH release, as the GH response is markedly impaired in patients with stalk agenesis.54
Endocrine Evaluation
The endocrine assessment of a patient with suspected hypopituitarism usually involves measurement of both baseline and stimulated hormone levels. Evaluation of baseline function involves prolactin, TSH, thyroxine (T4), cortisol, LH, FSH, and testosterone in men, and estradiol in women. Baseline blood testing reliably identifies hypothyroidism, hypogonadism, and severe hypoadrenalism due to pituitary insufficiency.
Dynamic Testing
Adult Gonadotropin Deficiency
In women of postmenopausal age, gonadotropin levels are clearly low or undetectable, whereas in premenopausal women, amenorrhea (or less commonly, oligomenorrhea), in addition to low estradiol levels and low or normal gonadotropin levels, provides sufficient evidence of the diagnosis. In adult men, a similar picture of low testosterone levels and low or inappropriately normal gonadotropin levels is seen.
Adrenocorticotropic Hormone Deficiency
A high index of clinical suspicion is most important in establishing the diagnosis. In normal people, the highest plasma cortisol levels are found between 6:00 am and 8:00 am, and the lowest before midnight. Plasma cortisol and ACTH concentrations are elevated during physical and emotional stress, including acute illness, trauma, surgery, infection, and starvation.
If a 9:00 am cortisol level is less than 100 nmol/L, particularly in an unwell patient, cortisol deficiency is highly likely, whereas a baseline level greater than 500 nmol/L indicates normality; many authors suggest that dynamic assessment of the hypothalamic-pituitary-adrenal (HPA) axis is not necessary under these circumstances.55 Unless the patient is known to have pituitary disease, a paired plasma ACTH level will help distinguish between primary and secondary glucocorticoid deficiency: In primary cortisol deficiency (Addison’s disease), the ACTH level will be high, whereas in secondary glucocorticoid deficiency, the ACTH level will be low or inappropriately normal.
If cortisol deficiency is suspected in an unwell patient, baseline cortisol and ACTH samples should be taken, and replacement therapy should be commenced immediately. Provocative testing can be performed at a later date.
The insulin tolerance test (ITT) is the test of choice in those suspected of secondary adrenal failure. The ITT evaluates the response of the HPA axis to the potent stressor of hypoglycemia, and it is generally the “gold standard” in the confirmation of secondary adrenal failure. It has the advantage of also being a test of growth hormone reserve in patients with pituitary disease.56 Following injection of a standard dose of intravenous insulin (0.1 unit/kg),57 cortisol concentrations are measured serially. Upon achievement of adequate hypoglycemia (<2.2 mmol/L), a peak cortisol response of between 500 and 600 nmol/L generally is accepted as adequate.58
The short Synacthen (tetracosactrin) test sometimes is used as a surrogate test of ACTH deficiency on the basis that the adrenal gland will respond to an exogenous bolus of synthetic ACTH when there is a normal endogenous ACTH reserve and the gland is not atrophic. Although it is a good test of adrenal reserve, it does not directly test pituitary ACTH reserve. In a patient with organic pituitary disease, a normal response to Synacthen does not exclude mild or recent ACTH deficiency.
Thyroid-Stimulating Hormone Deficiency
In secondary hypothyroidism, one might expect to find reduced concentrations of free or total T4 in association with a serum TSH concentration below the normal range, analogous to the biochemical findings in secondary hypogonadism. Most have normal or occasionally elevated TSH levels. The mechanism behind this apparent contradiction is poorly understood, but it may be due to reduced bioactivity of TSH,59 which suggests that TRH regulates not only the secretion of TSH but also its specific molecular and conformational features. Dynamic testing with thyrotropin-releasing hormone has little diagnostic value other than distinguishing a hypothalamic cause, which is indicated by a delayed TSH peak.
Antidiuretic Hormone Deficiency
The diagnosis of ADH deficiency first requires confirmation of polyuria, which is defined as the excretion of more than 3 L of urine per 24 hours (40 mL/kg/24 hours). Any patient with normal serum sodium and plasma osmolality who has a fluid output of <2 L/24 hours is likely to be normal and does not warrant further investigation.
Once excess urine output has been confirmed, the usual first-line investigation is an 8-hour fluid deprivation test. The test should be performed under strict observation because severe fluid and electrolyte depletion can occur. Plasma osmolality, urine volume, and osmolarity are measured hourly for 8 hours, after which a synthetic analogue of ADH (desmopressin) is given intramuscularly (IM). The urine osmolality then is remeasured. In a normal subject, ADH is secreted throughout the test, water is absorbed normally, and a subsequent elevation of urine osmolality occurs. In diabetes insipidus, the urine fails to concentrate (normal subjects achieve a urine osmolality at least twice the plasma osmolality) because of a lack of ADH; hence, plasma osmolality increases. Urine concentrates adequately only after administration of desmopressin. Sometimes in cases with long-standing polyuria, failure of urine concentration in response to desmopressin occurs not because of nephrogenic diabetes insipidus but because of a washout of interstitial solutes, including urea. This may lead to diagnostic difficulties. In cases in which the results of a water deprivation test are inconclusive, ADH measurement is helpful. A definitive diagnosis of ADH deficiency can be established by infusing hypertonic saline for 2 hours to increase plasma osmolality to more than 300 mOsm/kg, with regular 20 to 30 minute blood sampling to estimate plasma osmolality and ADH. In nephrogenic diabetes insipidus, ADH values are above the normal reference range, whereas in cranial diabetes insipidus, values are at the lower end of or below the normal reference range.
MANAGEMENT
Treatment for hypopituitarism can be separated into those therapies directed at the underlying disease process and endocrine replacement therapy (Table 13-4). Management of the underlying condition is particularly challenging for craniopharyngiomas because randomized studies are lacking and their growth pattern is often unpredictable.60 Surgical excision in combination with external beam irradiation forms the mainstay of treatment. A recent trial demonstrated no recurrence of tumors during an average follow-up of 6.5 years in patients who received combined surgery with radiotherapy at diagnosis. Sixty percent of patients who received surgery alone had tumor recurrence.61
Table 13-4. Endocrine Replacement Therapy for Hormone Deficiencies
| Hormone Deficiency | Replacement Hormones and Typical Daily Dose Range (Oral, if Not Stated Otherwise) |
|---|---|
| Growth hormone | Please refer to Table 13-7 in the section Growth Hormone Deficiency. |
| Gonadotropins (female) | Estrogen: |
| Estradiol valerate: 1-2 mg, transdermal: 25-100 µg | |
| or conjugated equineestrogens: 0.625-1.25 mg | |
| PLUS Progesterone (examples): | |
| Norethisterone, 0.7-1 mg, transdermal: 170-250 µg | |
| or Levonorgestrel, 250 µg, transdermal: 7 µg | |
| or Medroxyprogesterone acetate, 5 mg | |
| Gonadotropins (male) | Testosterone: |
| Intramuscular (as testosterone esters): 250 mg every 2-3 wk | |
| or Transdermal: 5-7.5 mg | |
| or Implant: 600-800 mg every 4-6 mo | |
| Thyroid-stimulating hormone | Thyroxine, 75-200 µg/day |
| Adrenocorticotropic hormone | Glucocorticoid (preferred schedule): Hydrocortisone, 10 mg morning, 5 mg noon, 5 mg evening, to 10 mg t.i.d. |
| Prolactin | Nil |
| Antidiuretic hormone | Desmopressin (DDAVP), 300-600 µg (in divided doses); intranasal, 10-40 µg (in divided doses) |
Hormone Replacement in Hypopituitarism
Endocrine replacement therapy should aim to mimic the normal hormonal milieu as far as possible, thus improving symptoms while avoiding overtreatment. GH replacement therapy is discussed separately under the section on adult GH deficiency.
Gonadotropin Deficiency
In both sexes, sex steroid replacement therapy is important for the maintenance of normal body composition, skeletal health, and sexual function, and it is the most appropriate form of replacement therapy in patients not desirous of fertility.
Estrogen Replacement
In women, this can be provided by many standard hormone replacement therapy preparations. Progesterone must be given (cyclically or continuously) in all women with an intact uterus to prevent the possible effect of unopposed estrogen on the endometrium, that is, dysfunctional bleeding or endometrial cancer. The dose of estrogen should not be supraphysiologic (as in the oral contraceptive pill) unless a clear indication, such as strong patient preference, exists, or a patient with partial gonadotropin deficiency still has occasional menstrual cycles, along with a desire for contraception. Although estrogen can be delivered as a tablet, patch, gel, or implant, a nonoral route is recommended because of reduction of insulin-like growth factor (IGF)-1 and fat oxidation by oral estrogen. The pathophysiology of the interaction of oral estrogen with the GH axis is discussed separately under the section on adult GH deficiency. However, an international surveillance study on 315 hypopituitary women taking estrogen replacement demonstrated significant predominance of oral versus transdermal estrogen use (86% vs. 14%). Women on oral estrogen had a significantly greater waist/hip ratio after GH treatment, with lower IGF-1 levels at the end of the study period on twice the GH dose received by women on transdermal estrogen.62 Therefore a nonoral route is highly recommended.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


