FIGURE 47-1. Hypoglycemia—Major barrier to intensive diabetes management.
(Data from Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–986.)
Death from hypoglycemia can be considered on the one hand to be rare when one considers the very large number of episodes of hypoglycemia that occur in clinical practice. However, mortality does occur during severe hypoglycemia in both type 1 and type 2 DM. Studies investigating the cause of death in patients with type 1 DM have reported that 2% to 10% may have died because of hypoglycemia.2,3 Similar death rates from hypoglycemia have been reported in patients with type 2 DM. The specific mechanism responsible for hypoglycemia-induced death is not currently understood. Possible suggested causes include cardiac arrhythmias, thrombotic events, and brain death. However, it should be noted that in primates, several hours of profoundly low glucose <1 mmol/L is required to induce irreversible brain damage and death.37
HYPOGLYCEMIA-ASSOCIATED NEUROENDOCRINE AND AUTONOMIC FAILURE
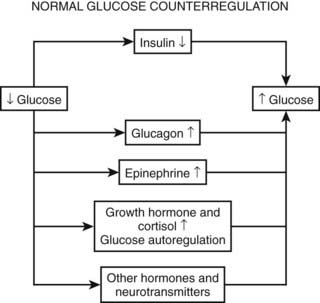
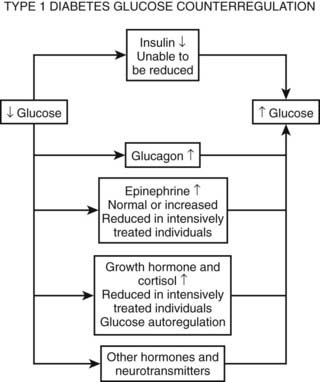
As discussed earlier, the four primary physiologic defenses against falling plasma glucose include inhibition of endogenous insulin secretion, release of glucagon and epinephrine, and symptomatic cues to ingest carbohydrate (Fig. 47-2). Unfortunately in patients with diabetes, all of these physiologic defenses can become defective and/or deficient. In type 1 DM and in long duration type 2 DM, the individual becomes critically insulin deficient, and thus the first physiologic line of defense (modulation of endogenous insulin) becomes lost. After varying periods of duration of disease (≈5 years), the ability of individuals with type 1 DM to release glucagon in response to hypoglycemia is lost (Fig. 47-3). This defect also occurs to a similar extent in long duration type 2 DM (Fig. 47-4).38 The mechanism for this finding is currently under intense investigation in humans. Hypotheses include lack of β cell turnoff, a possible specific autonomic nervous system dysfunction, and another as yet unidentified local signaling defect at the level of the α cell. The defect involved in releasing glucagon in type 1 DM is specific for hypoglycemia as the α cells in these individuals are present in normal number and size. In fact underscoring this point, glucagon responses to other metabolic stressors in type 1 DM such as exercise or amino acid infusions are preserved.39 The fact that glucagon responses are preserved during exercise in type 1 DM is interesting in that exercise is also typically associated with a physiologic “β cell switch off.” Whatever the mechanism, unfortunately after a few years’ duration, individuals with type 1 DM lose two of the four primary defenses against falling blood glucose. This leaves a functioning ANS (sympathoadrenal and sympathetic nervous systems) to serve as the primary defense against hypoglycemia in type 1 DM. In some individuals with type 1 DM, the ability to secrete epinephrine in response to hypoglycemia is preserved and can compensate for the lack of glucagon release.40 However, it is now clear that epinephrine responses to hypoglycemia are significantly reduced in patients with type 1 DM with intensive metabolic control.41 This epinephrine deficiency has been determined to be due to previous episodes of hypoglycemia42 and is separate from the syndrome of classic diabetic autonomic neuropathy that can occur after many years of suboptimal glycemic control. Models of repeated antecedent hypoglycemia have been demonstrated to produce acute reductions (30% to 50%) in epinephrine, pancreatic polypeptide (a marker of parasympathetic nervous system activity), and muscle sympathetic nerve activity (a direct marker of sympathetic nerve system activation) in individuals with type 1 DM, in those with type 2 DM, and in nondiabetic individuals.43 Additionally, recent (within 24 hours) antecedent hypoglycemia has been found to blunt a wide spectrum of neuroendocrine responses such as glucagon, growth hormone, ACTH, and cortisol during subsequent hypoglycemia44 in healthy and diabetic men. Confirming the role of antecedent hypoglycemia in causing blunted counterregulatory responses are a number of studies that prospectively investigated the effects of avoiding hypoglycemia in type 1 DM and following successful removal of an insulinoma.45–48 In all cases, there were initial blunted, neuroendocrine, ANS, and symptomatic responses to hypoglycemia. However, when patients with type 1 DM were restudied several months later, all showed improved counterregulatory responses to hypoglycemia, and patients with previous insulinomas had counterregulatory defenses restored to normal. Symptomatic responses, the fourth critical physiologic counterregulatory response, were significantly improved in all studies following a period of hypoglycemia avoidance.45–48 The blunting effects of antecedent hypoglycemia on subsequent counterregulatory responses have been termed by Cryer as “hypoglycemia-associated autonomic failure” (HAAF).
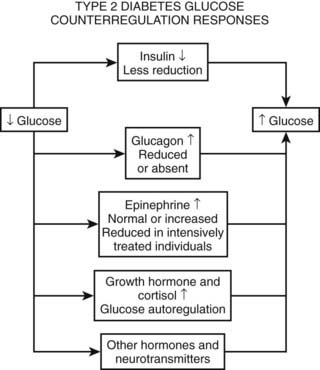
Following identification of this syndrome, a great deal of work has been performed in both animal and human models to further elucidate the mechanisms responsible for and the characteristics of hypoglycemia-associated counterregulatory failure. However, it should be appreciated that HAAF does not occur only in type 1 DM. Work from two independent laboratories has determined that this syndrome also occurs in type 2 DM.49,50 Segel and colleagues clearly demonstrated that moderate antecedent hypoglycemia of 50 mg/dL (2.8 mmol/L) can blunt ANS responses to subsequent hypoglycemia in moderately controlled (HbA1c 8.4%) individuals with type 2 DM.49 More recently, Davis and coworkers have demonstrated that even milder antecedent hypoglycemia of only 60 mg/dL (3.3 mmol/L) can blunt ANS responses to subsequent hypoglycemia in patients with type 2 DM with suboptimal (HbA1c ≈10.0%) or intensive glycemic control (HbA1c ≈6.7%).50
The great challenge in determining the mechanisms responsible for HAAF is explaining the simultaneous reduction in ANS-neuroendocrine responses and the change in glycemic thresholds that activate the physiologic defenses against falling plasma glucose. As described earlier, the usual physiologic thresholds for activation of ANS-neuroendocrine and symptom responses during hypoglycemia occur in the plasma glucose range between 50 and 80 mg/dL. In individuals with chronic hyperglycemia, symptoms of hypoglycemia can occur at plasma glucose levels between 90 and 140 mg/dL, depending upon the severity of the prevailing hyperglycemia. Simultaneous measurements of ANS-neuroendocrine hormones are in fact elevated during these symptoms, thus the individuals are experiencing the condition of “relative hypoglycemia.” This syndrome occurs because the thresholds for activation of ANS-neuroendocrine responses have been pushed to a higher plasma glucose level by the chronic hyperglycemia. On the other hand, individuals with intensive glucose control and multiple episodes of hypoglycemia often find that the activation of physiologic responses to hypoglycemia is pushed to a lower plasma glucose level. This dangerous condition, called hypoglycemic unawareness, results in inability of patients to recognize a falling plasma glucose until the value is below 50 mg/dL (2.9 mmol/L). In some individuals, a falling plasma glucose level is not recognized at plasma glucose levels of 30 mg/dL. This reduces the interval between first recognition of hypoglycemia and the onset of serious sequelae (such as coma or seizure). Thus, thresholds for the activation of physiologic defenses against hypoglycemia are labile and can change rapidly. The duration and depth of antecedent hypoglycemia required to induce HAAF have been characterized. Repeated episodes or relatively mild (3.9 mmol/L, or 70 mg/dL) and only brief durations (15 to 20 minutes) of hypoglycemia can independently blunt counterregulatory responses to subsequent hypoglycemia.51 However, one prolonged episode (2 hours) of moderate hypoglycemia (50 mg/dL, or 2.9 mmol/L) is sufficient to induce HAAF within a few hours on the same day.52
Numerous mechanisms responsible for the syndrome of HAAF have been proposed over recent years, with data both supporting and at times contrary to any given hypothesis. Boyle et al. proposed that repeated hypoglycemia increased cerebral glucose uptake in both healthy individuals and patients with type 1 DM, thereby reducing the stimulus for neuroendocrine counterregulatory responses during subsequent hypoglycemia.53 This finding was later challenged by work reporting no increase in blood-to-brain glucose transport following antecedent hypoglycemia.54 Other mechanisms that have been proposed include activation of the hypothalamic-pituitary-adrenal axis,44,55 increases in neurotransmitters such as GABA, and changes in hypothalamic fuel sensors such as glucokinase or AMP kinase (increases and decreases, respectively).56 Additionally, experimental evidence demonstrates that alcohol and opioids can downregulate subsequent ANS and neuroendocrine responses to hypoglycemia.57 Other physiologic mechanisms also have been found to cause forms of HAAF. These include sleep, exercise, and gender (Fig. 47-5).43,58 The association between exercise and hypoglycemia in type 1 DM has been both problematic and perplexing. Hypoglycemia can occur during, 1 to 2 hours after, or up to 21 hours after exercise. Traditionally, the explanation for this phenomenon was either a relative or absolute excess of subcutaneously injected insulin (due to an increase in insulin sensitivity following exercise) and/or incomplete glycogen repletion following exercise. Although these factors are important contributors to exercise-associated hypoglycemia, they cannot explain some of the profound episodes of hypoglycemia that occur during or after exercise. Studies from our own laboratory and others have demonstrated that exercise and hypoglycemia could reciprocally blunt subsequent ANS responses to either stress (Fig. 47-6).59,60 Thus, exercise blunts ANS responses (by 30% to 50%) to subsequent hypoglycemia, and vice versa. This feed-forward vicious cycle of blunted ANS responses between exercise and hypoglycemia can occur after only a few hours and persists for at least 24 hours following either stress (Fig. 47-7).39 Appreciation that deficient counterregulatory responses are also involved in the pathogenesis of exercise-related hypoglycemia explains why this phenomenon can occur many hours after exercise and provides a platform for therapeutic intervention. In general, in an individual with type 1 DM who is experiencing exercise-related hypoglycemia, it is appropriate to consider reducing both basal and mealtime insulin doses. Additionally, slightly raising glycemic targets and ensuring adequate carbohydrate repletion of glycogen stores are useful recommendations.
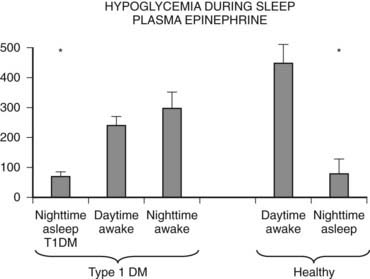
FIGURE 47-5. Hypoglycemia and plasma epinephrine responses during sleep/plasma epinephrine.
(Data from Jones TW, et al. N Engl J Med 1998;338:1657–1662.)
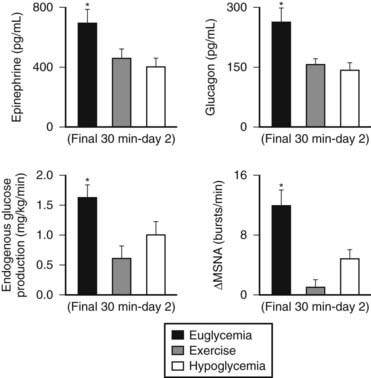
FIGURE 47-6. Reciprocal effects of antecedent hypoglycemia and exercise on subsequent neuroendocrine, autonomic nervous system, and metabolic (endogenous glucose production) responses to either stress.
(Data from Galassetti P, et al. Am J Physiol Endocrinol Metab 2001;280:E908–E917; Davis SN, et al. Diabetes 1997;46:1328–1335.)
Gender can also play a large role in modulating ANS and neuroendocrine responses during hypoglycemia.58 In premenopausal, nondiabetic, and type 1 DM women, moderate hypoglycemia of ≈50 mg/dL (2.9 mmol/L) produces 30% to 50% reduced ANS responses compared with that seen in age- and body mass index (BMI)-matched men.58 However, when postmenopausal women, not on estrogen replacement, were compared with postmenopausal women receiving estrogen and age- and BMI-matched men, it was found that the large sexual dimorphism in ANS (epinephrine, muscle sympathetic nerve activity) and neuroendocrine (glucagon, growth hormone) responses was no longer present in estrogen-deficient women. It therefore would appear that in healthy humans, estrogen is a major mechanism responsible for sexual dimorphic counterregulatory responses during hypoglycemia.61 It should also be mentioned that women appear to be more resistant than men to the blunting effects of prior hypoglycemia. Thus, antecedent hypoglycemia can have up to a twofold greater suppressive effect on subsequent counterregulatory responses in men than in women.62 The mechanism for this intriguing finding is as yet unknown. Many studies have focused on the blunting effects of antecedent hypoglycemia on neuroendocrine and symptom responses during subsequent hypoglycemia. However, an additional component in the spectrum of deficient counterregulatory responses deserves mention. Several laboratories have identified that adrenergic receptors and particularly epinephrine action are downregulated by intensive glucose control and prior hypoglycemia.63,64 These reduced metabolic (lipolytic, endogenous glucose production, glycogenolysis) and cardiovascular responses contribute to defective counterregulatory defenses against a falling plasma glucose in patients with diabetes. Thus, it should be noted that strategies aimed at increasing epinephrine levels during hypoglycemia will be only partially successful if tissue resistance or adrenoreceptor downregulation to the action of the hormone is present. What is clear from the above wealth of data is that multiple mechanisms can downregulate ANS responses to hypoglycemia, and that this complex model presents numerous targets for therapeutic interventions to stimulate and restore counterregulatory responses during hypoglycemia in patients with diabetes.
STRATEGIES TO IMPROVE COUNTERREGULATORY RESPONSES DURING HYPOGLYCEMIA
Parallel with studies investigating the mechanisms responsible for HAAF, a number of laboratories have been exploring strategies for improving ANS and neuroendocrine responses during hypoglycemia. These have included preclinical approaches in rodents through to interventions in humans with type 1 DM. As mentioned above, hypothalamic kinases can act as important fuel sensors. Recent work in rats has shown that methods to reduce AMP-activated protein kinase (AMPK) in the ventral medial nucleus of the hypothalamus reduce epinephrine and glucagon responses during hypoglycemia, whereas increases in AMP kinase action can increase responses of these key neuroendocrine hormones during hypoglycemia.65 Other approaches to increase neuroendocrine responses during hypoglycemia in man have included amino acid infusions (glucagon) and use of caffeine (epinephrine and symptom responses). Terbutaline before bed has been demonstrated recently to increase plasma glucose levels during the night and to prevent nocturnal hypoglycemia.66 Recently, peroxisome-proliferator–activated receptor-γ (PPAR-γ) agonists, which are known to activate AMP kinase and fructose, which under certain conditions can inhibit glucokinase,67 have been demonstrated to increase counterregulatory responses in both healthy men and those with type 1 DM. Although at first glance these latter studies may seem unrelated, both may have mechanisms working through hypothalamic fuel sensing. Most recently, studies in conscious rats and healthy and type 1 DM men have highlighted the possible role of serotonergic transmission in modulating counterregulatory responses during hypoglycemia. Prolonged (i.e., weeks) use of two different selective serotonin reuptake inhibitors (sertraline and fluoxetine) has led to dramatic (30% to 60%) increases in ANS (epinephrine) responses during hypoglycemia.68
DRUG-INDUCED HYPOGLYCEMIA
By far the most common cause of drug-induced hypoglycemia is insulin followed by sulfonylurea, meglitinides, and benzoic acid derivatives (i.e., oral insulin-producing agents). Hypoglycemia induced by oral insulin secretagogues is much less frequent than that caused by insulin but in certain instances can still be common. For example, hypoglycemia can occur in up to ≈35% of patients receiving glyburide or repaglinide. Hypoglycemia rates are less in agents with glucose-dependent insulin secretion (i.e., stimulation of insulin release is reduced during periods of hypoglycemia). Thus, the percentage of patients who experience hypoglycemia when receiving glimepiride, glipizide XL, or nateglinide is lower and is in the range of 5% to 10%. However, combination of even glucose-dependent insulin secretagogues with agents such as insulin or glucagon-like peptide-1 agonists can result in a much higher frequency of hypoglycemia (≥35%). Generally, newer sulfonylureas produce less hypoglycemia than do older first-generation sulfonylureas such as chlorpropamide. Severe hypoglycemia remains relatively uncommon with oral insulin secretagogues at a rate of ≈1.5 per 100 patient-years.33 Alcohol might be a more common cause of severe hypoglycemia in the United States than sulfonylureas.69 Alcohol can cause hypoglycemia in overnight fasted normal volunteers,70,71 with plasma glucose values as low as 5 mg/dL (0.3 mmol/L)72 and mortality rates ranging from 10% in adults to 25% in children.119 In a series of deaths caused by hypoglycemia, alcohol was the most common causative agent.74 The most common situation is a glycogen-depleted state, such as occurs in an individual who drinks after a considerable fast, or who drinks and then fasts. In the latter situation, blood alcohol levels can be low or undetectable.
Alcohol induces hypoglycemia by inhibiting gluconeogenesis72; as little as 50 g might be sufficient.73,76 Its mechanism of action is complex, with evidence of impaired counterregulatory hormone responses71 and impaired uptake of gluconeogenic precursors,77 but the predominantly accepted mechanism is its inhibition of the gluconeogenic process stemming from an increased reduced nicotinamide adenine dinucleotide (NADH)/NAD ratio as a result of the oxidation of alcohol to acetaldehyde and acetate, thus reducing the ability of the liver and kidney to oxidize lactate and glutamate to pyruvate and α-ketoglutarate, respectively.78–80 Although plasma insulin levels are appropriately suppressed in this condition, because of this inhibition of gluconeogenesis, glucagon and catecholamines are ineffective in increasing glucose release and raising plasma glucose levels.81 Thus, in a patient with suspected alcohol-induced hypoglycemia, oral or intravenous glucose is the treatment of choice.
Only about 10% of reported cases of drug-induced hypoglycemia have occurred without concomitant insulin, sulfonylurea, or alcohol.82 Of these, propranolol,83 sulfonamides,84 and salicylates169 have been reported most frequently. Propranolol and other nonselective β-blockers decrease the ability of the liver and kidney to increase their release of glucose,85,86 enhance peripheral insulin sensitivity,87 and mask symptoms of impending hypoglycemia. The adverse effects of β-adrenergic β-blockers are mediated through β2-receptors. Recent studies indicate that β1-selective blockers do not present an increased risk for severe hypoglycemia and therefore should not be considered as being contraindicated in diabetic patients.87,88
Salicylates can act by inhibiting hepatic glucose release and increasing insulin secretion, although their exact mechanism remains to be determined. Sulfonamides probably act by stimulating insulin release in a manner similar to that of sulfonylureas. Angiotensin-converting enzyme inhibitors89and pentamidine90 are associated more frequently with hypoglycemia, as their use increases in diabetic patients and those with AIDS, respectively. Angiotensin-converting enzyme inhibitors can increase tissue insulin sensitivity91 and can decrease the degradation of bradykinin, which has certain insulin-mimetic actions.92 Pentamidine is cytotoxic to pancreatic β cells, and hypoglycemia occurs with the release of insulin from degenerating cells, often with subsequent permanent diabetes mellitus.93 Many of the drugs listed in Table 47-2 have been reported to cause hypoglycemia only in association with the use of antidiabetic medications or have been the subject of isolated case reports, and their etiologic significance remains to be established. However, their use in a patient with otherwise unexplained hypoglycemia should be discontinued whenever possible.
TREATMENT AND STRATEGIES TO REDUCE HYPOGLYCEMIA
Clinical Strategies to Reduce Hypoglycemia
Over the last generation, interest has increased in replacing insulin in the most physiologic manner for patients with diabetes. In the 1980s, the introduction of recombinant human insulin reduced the formation of antibodies and provided more predictable pharmacokinetic profiles. The next decade produced analogue insulins that initially were designed to provide a quicker onset and a shorter duration of action. These insulins (lispro, aspart, glulisine) were designed to reproduce more closely the typical physiologic prandial spikes of insulin observed following meals. The second wave produced long-acting basal types of insulin (glargine, detemir) designed to mimic the background constitutive release of the hormone that regulates nocturnal and interprandial glycemia. Studies in type 1 DM have demonstrated that hypoglycemia (particularly nocturnal) can be reduced when short-acting analogues versus regular (soluble) insulin are used.94,96 Similarly, long-acting analogues have been demonstrated to reduce hypoglycemia by 20% to 33% in patients with type 2 DM when compared with NPH-based regimens.34 Thus, current recommendations are to use analogue-based insulin replacement regimens whenever possible.
Insulin pump development began in the 1970s and over the past 20 years has become a major method of insulin replacement in the United States. Although widely acknowledged for its usefulness, the cost of treatment (pump and supplies) has restricted a more widespread acceptance of this technology. Nevertheless, studies in children94 and pregnant women have demonstrated a reduction in hypoglycemia when compared with multiple daily insulin injection regimens.
Most recently, continuous, real-time glucose monitoring has been introduced into clinical practice. Currently, a variety of devices can be worn in combination with an insulin pump or independently. In a multicenter, randomized, controlled trial, the use of a continuous glucose monitor has been shown to reduce both glycosylated hemoglobin (HbA1c) and, as a secondary end point, the incidence of severe hypoglycemia in type 1 DM adults and children but not adolescents95 when compared with conventional self-blood glucose monitoring. Currently, another large randomized study is under way that will test the hypothesis that a continuous glucose sensor–driven insulin pump replacement approach will provide better HbA1c and less hypoglycemia than multiple daily injections of insulin and traditional self-blood glucose monitoring in both children and adults with type 1 DM.
Pancreas Transplantation and Hypoglycemia
Pancreas transplantation has been performed in patients with type 1 DM for over 25 years. Typically, patients have experienced episodes of severe hypoglycemia, and this is one of the major criteria for consideration of pancreas transplantation. Generally, rates of hypoglycemia improve dramatically in the first year after transplantation. However, ≈30% of patients report episodes of minor hypoglycemia. Most (but not all) studies also demonstrate that counterregulatory defenses are improved after pancreatic transplantation.97–99 Most notably, glucagon responses to hypoglycemia increase, accompanied at an early stage by some improvement in epinephrine and symptomatic responses. Improvements in glucagon responses appear to be persistent and have been documented in long-standing pancreas transplantation recipients of up to 19 years post transplant.100 Although the improvement in glucagon responses to hypoglycemia appears to be durable after long-term pancreatic transplantation, it is clear that improvements in epinephrine and symptomatic responses to hypoglycemia are not sustained.100 Along similar lines, it has been reported that physiologic insulin and glucagon responses during exercise are maintained for the most part following pancreas transplantation.101 Robertson and colleagues have investigated whether living donors of pancreas segments have normal counterregulatory responses to hypoglycemia.102 This has particular relevance, as ≈25% of donors can develop diabetes within 1 year after the procedure. It is interesting to note that despite deficient insulin and glucagon responses to glucose or arginine infusion, it was found that glucagon responses during hypoglycemia were preserved among donors.
A history of recurrent severe hypoglycemic events is also a major indication for islet cell transplantation. Although the number of transplanted individuals who are insulin independent is low (≈10%) up to 5 years after transplantation,103 it generally is reported that major episodes of hypoglycemia are significantly improved following islet cell transplantation.104 This finding appears to be due to the fact that there is some restoration of the ability to modulate insulin levels during hypoglycemia, as no reports have described significant increases in glucagons during hypoglycemia following islet cell transplantation.105 These findings underscore the importance of modulating insulin in the defense against hypoglycemia, but they also pose the intriguing question as to why there is an absent glucagon response. One plausible explanation is that the intrahepatic site for the islet transplant is the cause of the deficient glucagon response to hypoglycemia.106 In a recent series of elegant experiments in rats, Zhou et al.106 demonstrated that in the normal postprandial state, glucagon responses to hypoglycemia were absent in intrahepatially transplanted islets. However, when the animals were fasted for 48 hours and thus intrahepatic glucose flux was reduced, normal glucagon responses to hypoglycemia were obtained in the islet-transplanted animals.106 Supporting the theory that placing islets in the liver may be cause for the absent glucagon response during hypoglycemia are data indicating that alternate sites for islet cell transplantation away from the liver may produce improved glucagon responses to hypoglycemia.107
Hypoglycemia and Gastric Bypass Surgery
Accompanying the rapid increase in obesity is an increase in the number of bariatric surgical procedures performed. Recently, accumulating reports are demonstrating an increase in severe postprandial hypoglycemia following gastric bypass surgery.108–110 Although the disorder was initially thought to represent a version of the adult nesidioblastosis syndrome, subsequent analysis of the pancreata have revealed potentially alternative pathologic causes.111 It is interesting to that that there is a strong female preponderance, and this condition can develop months or even years after bypass surgery is performed. Treatment for hypoglycemia has proved to be problematic. Neither acarbose given to try to alleviate dumping syndrome symptoms by slowing glucose absorption nor somatostatin given to inhibit endogenous insulin secretion has been successful.112 Consequently, pancreatic resection was needed to reduce the occurrence of hypoglycemia in the six patients originally reported with this syndrome.108 Subsequent reports have highlighted that patients with this syndrome have exaggerated glucagon-like peptide-1 (GLP-1) and insulin responses to a mixed meal.113 In late 2007, Vella and Service provided an update on the Mayo Clinic’s experience regarding hypoglycemia following gastric bypass surgery.112 They reported that 43 patients required an ≈60% gradient-guided pancreatic surgical resection to alleviate hypoglycemia. Pathologic inspection revealed that most of the pancreata had islet hypertrophy with nesidioblastosis, although some also had one or more insulinomas. It appears that this syndrome of hypoglycemia is specifically associated with bypass surgery, as a recent report of gastric banding to induce weight loss did not report any significant hypoglycemia over a 2-year postprocedure follow-up period.114
NEONATAL HYPOGLYCEMIA
Plasma glucose levels below 70 mg/dL (3.9 mmol/L) commonly occur in newborn infants. It has been estimated that up to 50% of neonates can have plasma glucose levels below 50 mg/dL (2.9 mmol/L) following short-term fasting (up to 8 hours) after birth.115 Over the first 72 hours of life, gluconeogenic pathways mature, and the risk of hypoglycemia in normal infants is removed. However, in addition to the above physiologic causes of neonatal hypoglycemia, numerous pathologic conditions can cause hypoglycemia (Table 47-3).
Table 47-3. Causes of Neonatal Hypoglycemia
The most common causes of persistent neonatal hypoglycemia are the congenital hyperinsulinism syndromes. These syndromes have been estimated to occur at a rate of about 1:30,000 births worldwide. However, in certain areas (e.g., Finland, the Arabian Peninsula), these conditions have been reported to occur at much higher rates (≈1:2,100).116,117 Over the past 50 years, congenital hyperinsulinism syndromes have been given a number of titles (e.g., nesidioblastosis, islet dysregulation syndrome, persistent hyperinsulinemic hypoglycemia of infancy). Recently, it has been discovered that nesiodioblastosis can be a normal feature of the pancreas in the neonatal period and is histologically different from the pathologic processes that cause congenital hyperinsulinism. The pathophysiology and the molecular and genetic bases for congenital hyperinsulinism were reviewed recently by De Leon and Stanley.115 Of these, the most frequently occurring involve loss-of-function mutations in the pancreatic β-cell K-ATP channel, Kir6.2 and Sur-1 receptors.
The clinical presentation of infants with congenital hyperinsulinism may be varied but typically involves prolonged and severe hypoglycemia with lethargy, seizures, and apnea, and babies are often large for gestational dates. Typically, hypoglycemia occurs in the fasting or postprandial state, and these infants require large rates of glucose infusion >10 mg/kg/min to prevent severe hypoglycemia. In the glutamate dehydrogenase version of congenital hyperinsulinism, hypoglycemia can occur during both the fasting and the postprandial state. Hypoglycemia in this condition can be precipitated by a protein meal and is characteristically accompanied by elevated ammonia levels. Diagnosis of glutamate dehydrogenase hyperinsulinism typically occurs when the child is several months of age.118
Diagnostic Investigation of an Infant With Persistent Hypoglycemia
Because of the wide range of conditions that can cause hypoglycemia in an infant, a variety of blood and urine tests can be helpful in identifying metabolic and endocrine causes of the disorder (Table 47-4).
Table 47-4. Blood and Urine Tests for Metabolic and Endocrine Causes of Neonatal Hypoglycemia
Blood glucose • Insulin, C-peptide, proinsulin : insulin ratio, growth hormone, cortisol, thyroid hormones, thyroid-stimulating hormone (TSH) |
It is surprising that plasma insulin levels often are not strikingly elevated in the congenital hyperinsulinism syndromes. Similar to insulinomas, plasma insulin levels often are modestly increased but are inappropriately raised in relation to the prevailing hypoglycemia. However, measurement of free fatty acids and β-hydroxybutyrate levels reveals dramatic suppression and evidence of significant insulin action. An increase in plasma glucose greater than 30 mg/dL following glucagon is also supportive of the diagnosis. Infants with the GDH-M1 form of congenital hyperinsulinism exhibit increased responses to leucine118 and thus can be distinguished from those with the KATP channel forms of hyperinsulinism. Additionally, genetic testing is now widely available for identification of four of the five genes associated with congenital hyperinsulinism. Initially, treatment consists of diazoxide, which is a KATP channel agonist and has a suppressive affect on insulin secretion. If diazoxide does not produce a clinical response, octreotide has been used to suppress insulin release. However, in children who are unresponsive to medical therapy, the next treatment is surgery. This can comprise a local resection or a near total (98%) pancreatectomy, depending upon the presence of focal or diffuse disease.115,118 The application of 18F-DOPA positron emission tomography (PET) scans to differentiate focal from diffuse pancreatic involvement in the congenital hyperinsulinism syndromes has provided a clinical breakthrough in the management of these children.119
MITOCHONDRIAL FATTY ACID OXIDATION DISORDERS
Eleven different mitochondrial fatty acid oxidation disorders120 can present with fasting (hypoketotic) or postprandial hypoglycemia. Hepatic oxidation of fatty acids produces energy and acetyl-CoA, both of which are essential for gluconeogenesis. Additionally, fatty acid oxidation is required for hepatic ketone production. However, because of specific defects in mitochondrial fatty acid oxidation, gluconeogenesis is reduced, and this results in hypoglycemia and a lack of ketone bodies. The combination creates a scenario whereby the brain becomes starved of glucose and its primary alternative substrate (ketones), which creates conditions for severe neurologic consequences. Plasma membrane carnitine transporter deficiency is an autosomal recessive disorder that occurs in ≈1:40,000 infants. Low plasma carnitine and acyl carnitine are indicative of this disorder, and treatment with dietary carnitine prevents hypoglycemia. Carnitine palmitoyl transferase I and II (CPT I and CPT II) deficiencies can present as severe, life-threatening events that require frequent feeding and supplementation with dietary medium-chain triglycerides and l-carnitine (CPT II deficiency). Unfortunately, the most severe form of CPT II deficiency can present soon after birth and can be fatal. Medium-chain acetyl-CoA dehydrogenase (MCAD) deficiency is the most common fatty acid oxidation defect and is inherited as an autosomal recessive trait. Hypoglycemia usually occurs during fasting and conditions of stress such as viral illness.121,122 The presentation can be serious with vomiting, apnea, coma, encephalopathy, and death. It has been estimated that if left undiagnosed, up to 25% of children may die during the presenting event. Blood tests demonstrate elevated medium-chain acylcarnitine. As with other mitochondrial fatty acid oxidation disorders, the enzyme deficiency can be demonstrated diagnostically in fibroblasts. Treatment consists of avoidance of fasting and precipitation of attacks that could lead to neurologic defects and even death. Thus, frequent meals, bedtime snacks, uncooked cornstarch (to avoid nocturnal hypoglycemia), and carnitine supplementation have been used successfully in these children.
Very long chain (VLCAD) and short-chain acyl-CoA dehydrogenase (SCAD) deficiencies can present with serious, sometimes fatal, episodes of hypoglycemia and metabolic acidosis. These defects are also associated with cardiomyopathy (VLCAD), hepatomegaly, and neurologic defects (SCAD). Treatment involves avoidance of fasting and maintenance of glucose levels during stress, with frequent meals enriched with carbohydrate and medium-chain triglycerides. Carnitine supplementation in these conditions is not recommended (VLCAD) or has limited applicability (SCAD).
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) and mitochondrial trifunctional protein (MTP) deficiency are also autosomal recessive disorders. Both conditions can present with severe hypoglycemia, encephalopathy, neurologic complications, and death. Both conditions can result in peripheral neuropathy, and LCHAD deficiency can result in pigmentary retinopathy and blindness. Treatment is similar to that provided for the above fatty acid oxidation defects acyl-CoA dehydrogenase and carnitine/acylcarnitine translocase (CACT). Long-chain ketothilase (LCKAT) and 2-4-dienoyl-CoA reductase deficiency are very rare conditions that have been described recently.120
GLYCOGEN STORAGE DISEASES
Glycogen synthase and glucose-6-phosphatase deficiencies often present in the neonatal period with hypoglycemia. Additionally, glycogen-phosphorylase deficiency can present later in childhood. Of the three disorders, glucose-6-phosphate deficiency presents with the most severe hypoglycemic phenotype. Treatment to prevent hypoglycemia is similar in all of the glycogen storage disorders.123 Avoidance of hypoglycemia during stress, exercise, and the night involves frequent carbohydrate-containing meals and bedtime snacks. In times of severe stress, intravenous glucose may be required to maintain plasma glucose levels above 70 mg/dL (3.9 mmol/L).
SEPSIS, TRAUMA, AND BURNS
Initially, the response to the stress of infection is an increase in glucose turnover, with glucose production often exceeding glucose utilization and resulting in mild hyperglycemia. This response involves increases in both glycogenolysis and gluconeogenesis and is largely mediated by glucagon124 because adrenergic blockade has no effect on glucose turnover.125 As the infection worsens, increased release of endotoxin and its derivatives, complement activation, endoperoxide activation, and release of endogenous inflammatory mediators (tumor necrosis factor-α, interleukins, and other monokines) compromise cardiovascular integrity and cause central venous pooling, inadequate tissue perfusion, and microvascular protein transudation.126 At this stage, a decrease in splanchnic and renal blood flow occurs. Despite concomitantly reduced peripheral tissue perfusion, glucose utilization is increased.127–129 Decreased tissue oxygenation causes increased anaerobic glycolysis, which perpetuates the increased glucose utilization.
The inability of glucose release to keep pace with increased tissue demands results in hypoglycemia. Hepatic glycogen stores are rapidly exhausted; consequently, glucose release becomes solely dependent on gluconeogenesis. However, gluconeogenesis fails to stimulate this process because of a reduction in ANS and neuroendocrine effects.130–132 Factors such as acidosis (which inhibits hepatic gluconeogenesis), increased intracellular calcium (which impairs mitochondrial function and inhibits gluconeogenic enzymes), and siphoning of available energy from gluconeogenesis to support ion transport might be the mechanisms responsible for diminished ANS and neuroendocrine responsiveness.
Appropriate management entails (1) treatment of the underlying infection, (2) restoration of normal peripheral perfusion, and (3) glucose infusion to satisfy tissue demands.
CARDIAC FAILURE
Spontaneous hypoglycemia can occur with severe heart failure133–135; it is rare in adults but not uncommon in infants and children,136 in whom reduced hepatic glycogen levels (but normal phosphorylase and glucose-6-phosphatase activity) have been found in liver biopsy specimens. The condition has been attributed to a variety of mechanisms, including reduced gluconeogenesis, poor dietary intake, and gastrointestinal malabsorption, which are present in cardiac failure.
Mellinkoff and Tumulty first described the hypoglycemia of cardiac failure and attributed it to associated hepatic disease.137 However, chronic lung disease with right and left heart failure is seen in most patients.135 Thus, hypoxemia and low cardiac output may produce hepatic ischemia. Marks and Rose postulated that decreased availability of oxygen would suppress gluconeogenesis by increasing hepatic anaerobic glycolysis (Pasteur effect) and lactate production, thereby resulting in an increased NADH/NAD ratio.138 This decreased ratio could compromise gluconeogenesis because NAD is an essential cofactor for several of the enzymatic steps of gluconeogenesis. This attractive hypothesis could explain the association between hypoglycemia and the lactic acidosis of cardiac139 and liver140 disease, as well as the hypoglycemia accompanying other conditions associated with tissue anoxia, such as sepsis and shock.141 Low cardiac output would be expected to limit substrate delivery to the kidneys. In addition to a reduced capacity to produce glucose, increased glucose utilization from increased anaerobic glycolysis and increased energy demands from labored breathing and malnutrition (anorexia) are probably additional important factors. At the present time, no evidence indicates that abnormal counterregulatory hormone responses play a role in the pathogenesis of the hypoglycemia associated with these conditions.
RENAL AND HEPATIC DISEASE: GENERAL CONSIDERATIONS
The liver and kidneys are the only organs that are capable of releasing glucose into the circulation, inasmuch as other tissues generally lack or have minimal amounts of the enzyme glucose-6-phosphatase. Consequently, it would not be surprising that patients with hepatic or renal disease should be prone to hypoglycemia. Nevertheless, it is uncommon for hypoglycemia to occur simply as a result of loss of mass or function of these organs, and when it does occur, the cause is usually multifactorial.72,142 The large capacities of these organs to release glucose into the circulation and their ability to compensate for each other’s shortcomings appear to provide an explanation for this phenomenon.
Normally, the liver accounts for 80% to 85% of all glucose released into the circulation; it can increase its output (initially mainly by glycogenolysis, later by gluconeogenesis) over a sustained period by twofold to threefold (at least for several days, as exemplified by burn patients130). Thus, hypoglycemia with an appropriate compensatory increase in hepatic glucose release would be unlikely to develop in anephric individuals because the kidney normally contributes only 15% to 20% of all glucose that is released into the circulation. On the other hand, the kidney can increase its output over a prolonged period by twofold to threefold, as exemplified in humans who have fasted for several weeks.143 Animal and human studies indicate that the kidney can acutely increase its output to compensate for decreased hepatic glucose release and vice versa,144–146 a phenomenon that is referred to as hepatorenal reciprocity.147 For example, during the anhepatic phase of human liver transplantation, the kidney can maintain normoglycemia without the need for exogenous glucose.148 Thus, hypoglycemia would be unlikely to develop in patients with hepatic disease until the liver’s capacity to release glucose was reduced beyond the ability of the kidney to compensate. In fact, animal studies indicate that more than 80% of the liver must be removed for hypoglycemia to occur.149
Liver Disease
Although hypoglycemia has been associated with a wide range of liver diseases (hepatocellular carcinoma, cirrhosis, fatty metamorphosis, toxic and infectious hepatitis, cholangitis, and biliary obstruction), its occurrence is actually uncommon in the absence of other complicating factors (i.e., infection).72,138 For example, Zimmerman and coworkers found fasting hypoglycemia levels of 60 mg/dL (3.3 mmol/L) or less in only 6 of 269 patients with a variety of liver diseases.150 In humans, the insult to hepatic function must be acute, or the loss of parenchyma must be widespread. Felig and coworkers found that about 25% (4 of 15) of patients with acute viral hepatitis had a fasting blood glucose level less than 50 mg/dL (2.8 mmol/L).151 In chronic liver disease associated with hypoglycemia, additional factors, such as malnutrition and infection, usually are involved. A 50% incidence of hypoglycemia was found in patients with liver disease associated with sepsis and circulatory collapse.152,153 In such situations, liver function tests might not parallel the severity of hypoglycemia. Infiltrative diseases such as metastatic disease, amyloidosis, sarcoidosis, and hemachromatosis rarely replace sufficient parenchyma to cause hypoglycemia.154
Hyperinsulinemia often accompanies hepatic disease as a consequence of decreased insulin degradation by the liver,155 but the hypoglycemia of hepatic disease is almost always accompanied by appropriate suppression of plasma insulin concentrations.151 Likewise, little evidence has been found to implicate overproduction of insulin-like growth factors (IGFs) by the liver. Gorden and colleagues reported one patient with hemangiopericytoma of the liver in their series of 52 patients with extrapancreatic tumors, hypoglycemia, and IGFs, but no excessive IGFs were associated with primary hepatocellular carcinoma, the hepatic neoplasm most frequently associated with hypoglycemia.156
Renal Disease
Except in infants, hypoglycemia rarely occurs with acute renal failure. However, with chronic renal failure, hypoglycemia is not uncommon in adult patients. It can occur as an isolated event or can be repetitive. In general, neuroglycopenia rather than autonomic symptoms predominates.142
Although it has long been recognized that uremia reduced the insulin requirement in diabetic humans,157 it was not until 1970 that Block and Rubinstein reported three diabetic patients with renal failure who had suffered severe hypoglycemia after insulin and sulfonylurea therapy had been stopped.158 Shortly afterward, spontaneous hypoglycemia associated with renal failure was described in nondiabetic patients,159–162 with an incidence of 1% to 3% in two large studies.162,163
The origin of renal hypoglycemia is complex.142,164 Many factors, including altered drug metabolism, delayed gastric emptying, malnutrition, infection, dialysis, altered insulin sensitivity, associated hepatic and cardiac disease, and impaired renal and hepatic glucose release, predispose uremic patients to hypoglycemia. Drugs are probably the most common immediate cause. Any drug that can cause hypoglycemia is more likely to do so in a uremic patient because of a prolonged half-life (e.g., insulin, certain sulfonylureas, especially chlorpropamide) or decreased protein binding secondary to hypoalbuminuria.165 Although hypoglycemia occurs in nondiabetic as well as diabetic patients with renal failure, it is more likely to occur in the latter because of the use of hypoglycemic agents, and because patients with long-standing diabetes have autonomic neuropathy and defects in glucose counterregulation. Most patients have been malnourished, although the condition has been reported in well-nourished patients.160 Malnutrition secondary to anorexia or vomiting, which can reduce hepatic glycogen stores and the availability of gluconeogenic precursors, is a common feature that increases the risk for hypoglycemia.166
Fatal hypoglycemia can occur with peritoneal dialysis or hemodialysis when high glucose–containing dialysate is used, because of exaggerated insulin release in conjunction with impaired renal insulin degradation.167 Other factors such as the use of glucose-deficient solutions in diabetic patients and loss of alanine during hemodialysis may contribute to the development of hypoglycemia.168,169
Several reports suggest that renal failure per se predisposes to the development of hypoglycemia.158,161,162,170–176 Most evidence points to diminished glucose release rather than increased glucose utilization as a cause of the hypoglycemia.160,162,177–179 Accompanying malnutrition and acidosis would also contribute to diminished hepatic glycogenolytic and gluconeogenic potential.134,171,173,180–182 The expected compensatory increase in renal gluconeogenesis in response to acidosis would be compromised by loss of renal mass and exacerbated by inappropriate plasma insulin levels caused by reduced renal insulin degradation.
GLUCAGON DEFICIENCY
Pancreatectomized individuals, patients with long-standing type 1 diabetes, and those in whom insulin-requiring diabetes develops as a result of chronic pancreatitis or cystic fibrosis are glucagon deficient182 and prone to severe hypoglycemia during treatment for their diabetes.183 An imbalance between insulin and glucagon secretion (relative glucagon deficiency) has been reported in reactive postprandial hypoglycemia.184 Because of glucagon’s importance, it might have been expected to be a common counterregulatory hormone deficiency. However, the condition is extremely rare, with only two poorly substantiated cases of neonatal hypoglycemia185,186 and two reported cases in adults.187,188
CATECHOLAMINE DEFICIENCY
Patients with long-standing type 1 diabetes mellitus, adrenalectomized individuals, and those with autonomic neuropathy have impaired catecholamine responses during insulin-induced hypoglycemia,189–191 but their increased risk for hypoglycemia can be compensated for by increases in the secretion of other counterregulatory hormones, in particular, glucagon.192 Subtle defects in recovery from hypoglycemia have been demonstrated when such compensatory increases have been prevented experimentally.394 However, if glucagon responses to hypoglycemia are impaired simultaneously (e.g., in the patient with type 1 DM), the risk for hypoglycemia markedly increases.183
Several cases of neonatal ketotic hypoglycemia and one case in a 5-year-old boy have been attributed to epinephrine deficiency on the basis of low urinary epinephrine excretion.193,194 No cases of hypoglycemia secondary to isolated catecholamine deficiency have been reported in an adult. The hypoglycemia that occurs with propranolol may relate to the drug’s inhibition of lipolysis, which would reduce gluconeogenesis, an important counterregulatory process, and its promotion of increased glucose clearance by peripheral tissues.85 It is important to note that acute hypoglycemia can occur during surgical removal of a pheochromocytoma, presumably because of disinhibition of insulin release and abrupt withdrawal of the anti-insulin actions of catecholamines.195
CORTISOL AND GROWTH HORMONE DEFICIENCY
Although cortisol and growth hormone have been demonstrated to contribute independently to glucose counterregulation via their actions to promote glucose release and limit glucose uptake,196–199 hypoglycemia does not develop in most adults who lack these hormones. However, serious hypoglycemia often develops in infants and children, who lack these hormones, especially after a period of fasting or during an intercurrent illness.199–203 In a review of 76 adults with isolated adrenocorticotropic hormone (ACTH) deficiency,204 24 (≈33%) had hypoglycemia. During prolonged fasting (6 days), adult growth hormone–deficient dwarfs become hypoglycemic,205 as can hypopituitary pregnant women.206 On the other hand, overnight fasting plasma glucose levels have been reported to be normal in glucocorticoid-withdrawn patients with primary adrenal insufficiency207 or panhypopituitarism.208 Acute adrenal insufficiency such as in Sheehan’s syndrome can be manifested as severe hypoglycemia,209 and autoimmune Addison’s disease may be the cause of severe recurrent hypoglycemia in a patient with type 1 diabetes mellitus.210
It is important to be aware that malabsorption of glucocorticoids can occur in patients with bowel disease and in patients treated with drugs such as bile acid sequestrants.211
AUTOIMMUNE HYPOGLYCEMIA
Of the two types of autoimmune hypoglycemia, one is due to autoantibodies against the insulin receptor, and the other is due to autoantibodies against insulin itself in individuals who have never received exogenous insulin. Both are rare and can produce fasting, as well as postprandial reactive hypoglycemia—primarily the former.212–220
Anti-Insulin Receptor Antibodies
Fewer than 30 patients in whom hypoglycemia developed as a result of antibodies directed against the insulin receptor have been reported. These antibodies act as agonists and produce hypoglycemia similar to insulin. Most patients have had evidence of other conditions associated with altered autoimmunity (systemic lupus erythematosus, scleroderma, primary biliary cirrhosis, immune thrombocytopenic purpura, celiac disease, Hashimoto’s thyroiditis, and Hodgkin’s lymphoma).216 Some patients have developed severe insulin resistance as a result of the antibodies blocking the insulin receptor before the antibodies became agonists. Patients with this condition have low circulating insulin and C-peptide levels, normal IGF-1, and appropriate counterregulatory hormone responses.
Although experience is limited, antibody titers generally decrease over time, and remission eventually occurs in most patients. However, because of the severity of the hypoglycemia, aggressive treatment is indicated. High-dose glucocorticoids,221,222 plasmapheresis,223 and alkylating agents224 all have been tried with variable success.
ANTI-INSULIN ANTIBODY HYPOGLYCEMIA
Since 1970, approximately 200 cases of anti-insulin antibody hypoglycemia have been reported, nearly 90% occurring in Japanese patients.213 Associated autoimmune disorders and plasma cell dyscrasias (Graves’ disease, rheumatoid arthritis, polymyositis, and systemic lupus erythematosus) are common. The use of certain drugs (hydralazine, procainamide, penicillamine, interferon-α, and methimazole) has been implicated in initiating the syndrome.216
Postprandial hypoglycemia is more common with this syndrome than is fasting hypoglycemia. Circulating insulin levels are increased and C-peptide levels might not be suppressed, as they are in patients who are taking insulin surreptitiously. Hypoglycemia occurs because dissociation of insulin from the antibodies causes prolonged hyperinsulinemia. Unlike the other syndrome of autoimmune hypoglycemia, the course of this condition is benign and self-limited, with remission usually occurring within a year. Simple interventions such as frequent small meals with a low content of simple sugars often suffice.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


