FIGURE 132-1. Outline of the organization of the major steroid biosynthetic pathways in the adrenal cortex. The area within the dotted square contains the core steroidogenic pathways, of which the left column shows the Δ5-pathway and the right columns the Δ4-pathway, also utilized by ovarian theca cells. The top lightly shaded row shows the pathway to aldosterone; the middle row shows the zona fasciculata pathway to cortisol. The lower, darkly shaded row shows the zona reticularis steps to dehydroepiandrosterone sulfate (DHEA-SO4) and other 17-ketosteroids. Dotted pathways are considered to be relatively minor. Cmpd S = 11-deoxycortisol; the 11-deoxy intermediate to aldosterone (deoxycorticosterone) is not shown. The steroidogenic enzymes are italicized. Cytochrome P450 enzyme steps are: side-chain cleavage (scc); 17α-hydroxylase/17,20-lyase; 21-hydroxylase (21); 11β-hydroxylase/18-hydroxylase-dehydrogenase (11,18); and aromatase. Non-P450 enzyme steps are steroidogenic acute regulatory protein (StAR); Δ5-isomerase-3β-hydroxysteroid dehydrogenase (3β); 17β-hydroxysteroid dehydrogenase (17β); and sulfotransferase (ST). Clinically relevant electron transfer enzymes are P450 oxidoreductase (POR) and cytochrome b5 (b5).
(Modified with permission from Rosenfield RL: Identifying children at risk for polycystic ovary syndrome, J Clin Endocrinol Metab 92:787, 2007. © The Endocrine Society.)
The biosynthesis of androgens is modulated by two key cytochrome P450 enzymes: cytochrome P450scc (CYP11A) and cytochrome P450c17 (CYP-17). The former mediates the chemical reactions involved in cholesterol side-chain cleavage and, together with the cholesterol transport enzyme, steroidogenic acute regulatory protein (StAR), is the rate-determining step for the formation of all steroid hormones in both the adrenal glands and gonads.1–3 Cytochrome P450c17 is rate limiting for the formation of cortisol and sex hormones, and its level of expression is absolutely dependent upon trophic hormone stimulation. This one enzyme possesses both 17-hydroxylase and 17,20-lyase activities. The first of these two sequential activities is necessary to form cortisol, and both activities are necessary to form the 17-ketosteroids, dehydroepiandrosterone (DHEA) and androstenedione, which are in turn the precursors of all potent sex steroids. P450c17 mediates conversion of pregnenolone by a two-step chemical reaction involving 17-hydroxylation (to 17-hydroxypregnenolone) followed by 17,20-lyase activity (to DHEA). Progesterone undergoes parallel reactions: P450c17 carries out 17-hydroxylation to 17-hydroxyprogesterone (17-OHP), and this in turn is converted to androstenedione by 17,20-lyase activity. Whether P450c17 is the source of 17,20-lyase activity in this path is unclear. The 17,20-lyase activity of P450c17 is regulated differently from its 17-hydroxylase activity; phosphorylation of enzyme serine residues and electron-transfer enzymes up-regulate the lyase activity. Δ5-Isomerase-3β-hydroxysteroid dehydrogenase (3β-HSD) and 17β-HSD are non-P450 steroidogenic enzymes. 3β-HSD type II is required for the adrenal glands and gonads to convert pregnenolone to progesterone and DHEA to androstenedione. Androstenedione is the major precursor for both testosterone and estrogen synthesis; in the ovaries it is converted by 17β-HSD type V to form testosterone or aromatized by aromatase (cytochrome P450arom) to form estrone.
Under normal circumstances, the ovaries and adrenal glands contribute nearly equally to testosterone production (Fig. 132-2).4 Approximately half of the total testosterone originates from direct secretion, while half is derived from peripheral conversion of secreted 17-ketosteroids. Other tissues including liver, adipose tissue, and skin express the enzymes 3β-HSD type II, 17β-HSD type 5, and P450arom, which allow for the further processing of steroids synthesized within the adrenal and gonad.
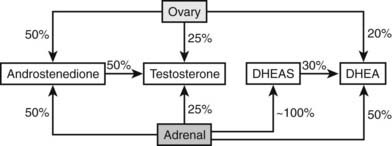
FIGURE 132-2. Relative contribution by the ovary and adrenal glands to circulating androgens and proandrogens in normal women. Approximately 25% of circulating testosterone is derived from the ovary; an equal proportion is of adrenal origin. The remaining 50% of testosterone in the circulation is derived from the conversion of androstenedione, which in turn is equally contributed to by the adrenal and ovary. The majority of DHEA is of adrenal origin: 50% is directly secreted, and 30% is derived from conversion of DHEAS. A small proportion (20%) of circulating DHEA comes from ovarian secretion. Finally, DHEAS is almost entirely derived from direct secretion by the adrenal.
(Data from Burger HG: Androgen production in women, Fertil Steril 77(Suppl 4):S3–5, 2002.)
REGULATION OF ANDROGEN SECRETION
Androgens are secreted by both the adrenal glands and ovaries in response to their respective trophic hormones, adrenocorticotropic hormone (ACTH) and luteinizing hormone (LH).5 Since androgens are in a sense byproducts of estradiol and cortisol secretion by the ovary and adrenal gland, respectively, androgen levels in females are not controlled by direct negative feedback by the pituitary gland, as is the case for cortisol and estradiol secretion. Rather, intraglandular paracrine and autocrine mechanisms seem to play a major role in the regulation of androgen secretion by these glands (see following and Table 132-1).
Table 132-1. Modulators of Theca Cell Androgen Biosynthesis
| Factor | Effect Upon Theca-Cell Androgen Biosynthesis | |
|---|---|---|
| Stimulation | Inhibition | |
| Autocrine | ||
| Transforming growth factor α | • | |
| Transforming growth factor β | • | |
| Hepatocyte growth factor | • | |
| Keratinocyte growth factor | • | |
| Nerve growth factor | • | |
| Paracrine | ||
| Transforming growth factor β | • | |
| Activin | • | |
| β-Fibroblast growth factor | • | |
| Tumor necrosis factor α | • | |
| Interleukin 1 β | • | |
| Bone morphogenetic protein 4 | • | |
| Insulin-like growth factor 1 | • | |
| Inhibin | • | |
| Growth differentiation factor 9 | • | |
| Endocrine | ||
| Luteinizing hormone | • | |
| Insulin | • | |
Adapted from Magoffin D: The role of the ovary in the genesis of hyperandrogenism. In Leung P, Adashi E (eds): The ovary, ed 2, San Diego, CA, 2003, Elsevier, pp 513–522.
Adrenal
Adrenal 17-ketosteroid secretion gradually begins during midchildhood as a result of adrenarche, the point at which there is a change in the pattern of the adrenal secretory response to ACTH. There are marked increases in 17-hydroxypregnenolone and DHEA production6 that lead to DHEA-sulfate (DHEAS, the sulfated derivative of DHEA) becoming the predominant androgen secreted by the adrenal gland. The conversion of DHEA to DHEAS is catalyzed by the sulfotransferase SULT2A1.
These adrenarchal changes seem to be related to the development of the zona reticularis of the adrenal cortex, an adrenal zone that produces large amounts of DHEA and DHEAS.5,6 This zone has low 3β-HSD activity and possesses the sulfokinase activity to sulfate the DHEA which accumulates. DHEA synthesis increases because of a combination of the low 3β-HSD activity and a change (of unknown origin) in regulation that includes increased expression of cytochrome b5, which enhances the 17,20-lyase activity of P450c17.
The nature of the factor or factors responsible for bringing about the adrenarchal change within the adrenal gland has been the subject of considerable controversy.5–7 Because ACTH levels do not increase in adrenarche but are necessary and “permissive” for its development,6 several substances have been implicated as the putative pituitary hormone responsible for bringing about the adrenarchal change, including “cortical androgen stimulating hormone,” pro-opiomelanocortin (POMC)-related peptides, and prolactin. The fact that adrenarche represents a change in the steroidogenic response pattern of the adrenal gland to ACTH suggests that potential adrenarche factor(s) may control the growth and differentiation of the zona reticularis and/or regulate steroidogenic enzyme activity, especially the 17,20-lyase activity of P450c17. Intraadrenal hormones such as estradiol,8 as well as extraadrenal hormones such as leptin, have also been postulated as potential adrenarchal factors.9,10 No single factor or hormone has yet been identified as both necessary and sufficient for the development of adrenarche.
Analogous to the case in the ovary, insulin and insulin-like growth factors (IGFs) may alter the response of adrenal steroids to ACTH.11 Insulin infusion has been reported to modestly potentiate the 17-ketosteroid response to ACTH in a pattern compatible with increases in 17-hydroxylase and 17,20-lyase activities, the former more prominently.12 Insulin excess might explain the enhanced responsiveness of 17-ketosteroids to ACTH in simple obesity. In vitro studies have directly shown that insulin and IGFs up-regulate adrenal 17-hydroxylase, 17,20-lyase, and 3β-HSD activities.
Ovary
The combined action of LH on theca-interstitial (thecal) cells and of follicle stimulating hormone (FSH) on ovarian granulosa cells, respectively, is necessary for the development and maintenance of normal ovarian function (Fig. 132-3).5 Androgenic precursors (especially androstenedione) are produced by thecal cells under LH stimulation. These are aromatized to estrogen by FSH-stimulated granulosa-cell aromatase activity. In the primate granulosa cell, proliferation and the growth of the dominant follicle is primarily dependent on FSH and FSH-induced growth factors.13 As the dominant follicle emerges, both increased amounts of androstenedione and estradiol are secreted, with the ratio favoring estradiol in healthy preovulatory follicles.
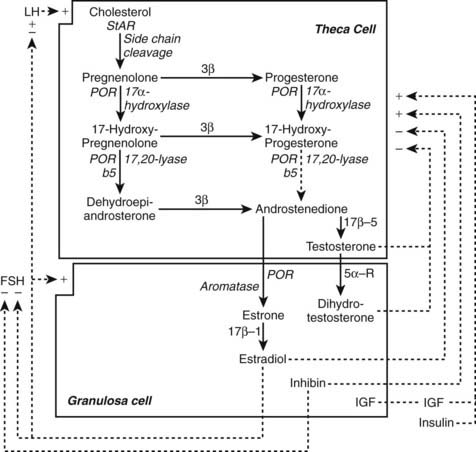
FIGURE 132-3. Outline of the organization of the major steroid biosynthetic pathways in the small antral follicle of the ovary, depicted according to the two-gonadotropin, two-cell model of ovarian steroidogenesis. LH stimulates androgen formation within theca cells via the steroidogenic pathway common to the gonads and adrenal glands. FSH regulates estradiol biosynthesis from androgen by granulosa cells. Long-loop negative feedback of estradiol on gonadotropin secretion does not readily suppress LH at physiologic levels of estradiol and stimulates LH under certain circumstances. Androgen formation in response to LH appears to be modulated by intraovarian feedback at the levels of 17-hydroxylase and 17,20-lyase, both of which are activities of cytochrome P450c17. The quantitative importance of androstenedione formation from 17-hydroxyprogesterone (dotted arrow) in the intact follicle is unknown. Androgens and estradiol inhibit (minus signs) and inhibin, insulin, and insulin-like growth factor 1 (IGF-1) stimulate (plus signs) 17-hydroxylase and 17,20-lyase activities. 3β, Δ5Isomerase-3β-hydroxysteroid dehydrogenase; 17β, 17β-reductase; 21, 21-hydroxylase; 5α-R, 5α-reductase; StAR, steroidogenic acute regulatory protein.
(Modified from Ehrmann D, Barnes R, Rosenfield R: Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism, Endocr Rev 16:322–353, 1995.)
Androgens are obligate intermediates in the biosynthesis of estradiol (see Fig. 132-3) and appear to have a dual role in follicular development within the ovary. Intraovarian androgens enhance the initial recruitment of primordial follicles into the growth pool and the growth of small antral follicles, while at the same time they appear to impair the cyclic recruitment of late antral follicles, thus leading to follicular atresia.14 Atretic follicles are relatively deficient in aromatase activity and thus contribute to androgen excess. Therefore, it seems critical for the function of the ovary that the intraovarian androgen concentration be kept at a level that is sufficient for the initial recruitment of primordial follicles and the production of estradiol, which is essential to prevent atresia. It is not known how ovarian androgen secretion is minimized while the formation of estrogen is optimized. Our studies suggest that androgen excess is normally prevented by the down-regulation process whereby excessive or protracted LH stimulation leads to intraovarian desensitization of the responses to LH, primarily at the level of P450c17.15
A number of hormones and growth factors seem to be involved in the intraovarian modulation of P450c17 steroidogenic responsiveness to LH (see Table 132-1 and Fig. 132-3).15,16 Desensitization to LH is mediated in part by ovarian steroids. It appears likely that estrogen inhibits this response by a short-loop (paracrine) negative-feedback mechanism. Androgens, on the other hand, may well be inhibitory by an autocrine mechanism. These inhibiting modulators seem to be counterbalanced by hormones and growth factors which amplify P450c17 activities, including insulin, IGF-1,17,18 and inhibin, as well as numerous other small peptides.5,16
BLOOD LEVELS AND TRANSPORT OF ANDROGENS
The total plasma concentrations of androgens and intermediates in their biosynthesis are shown in Table 132-2. Plasma levels of DHEAS normally reflect adrenal androgen production, while plasma testosterone and androstenedione levels are accounted for by both ovarian and adrenal androgen secretion. Plasma 17-OHP, like plasma testosterone and androstenedione, may arise from either the adrenal glands or ovaries. Of the circulating androgens, testosterone is the most important biologically and clinically because of its relatively high plasma concentration and potency at the target-organ level.

Over 96% of the plasma testosterone and structurally related 17β-hydroxysteroids circulate in plasma bound to carrier proteins, with only a small fraction remaining free.19,20 Sex hormone–binding globulin (SHBG) and albumin are the principal sex-steroid binding proteins in plasma. Although there has been interest in the possibility that albumin-bound testosterone is bioactive, most evidence suggests that it is only the free steroid intermediate that is bioavailable.21,22 Because of its high binding affinity, SHBG concentration is the major determinant of the fraction of 17β-hydroxysteroids binding to plasma albumin and of the fraction remaining free and biologically active in blood.
SHBG is a glycoprotein synthesized in the liver.20 Its synthesis is modulated by a number of physiologic and pathologic states that ultimately impact on its plasma concentration: SHBG levels are increased by estrogens and thyroid hormone excess; plasma concentrations are decreased by androgen, glucocorticoid, growth hormone, and insulin.23 Because SHBG levels are often decreased in hyperandrogenic states and obesity, serum free testosterone levels are often elevated in women whose total testosterone levels are normal. Consequently, the plasma free testosterone concentration is more often elevated in women with hirsutism or acne than is the plasma total testosterone concentration, reflecting the truly elevated plasma concentration of bioavailable androgen.24
MECHANISMS OF ANDROGEN ACTION
Testosterone exerts its effect by binding to the androgen receptor of target tissues. However, the biological activity of testosterone is dependent in large part on its being converted to dihydrotestosterone (DHT) by 5α-reductase. The formation of DHT from testosterone and other precursors determines, to a significant extent, the expression of androgen action at the level of the pilosebaceous unit.25,26 DHT is primarily formed by the 5α-reductase activity of target tissues and liver.27 In skin, 5α-reductase activity resides primarily in sebaceous and sweat glands, as well as dermis, rather than in hair itself. Measurement of plasma or urinary 3α-androstanediol glucuronide, a metabolite of DHT, has been touted as a marker of hypersensitivity of the hair follicle to androgen.28 However, the extent to which this metabolite arises from hepatic or skin 5α-reductase activity is controversial.29 DHT has greater biological potency than testosterone, owing to its higher affinity for and slower dissociation from the androgen receptor.30 The length of the androgen receptor’s CAG repeat, a polymorphism in exon 1, is potentially inversely related to the transactivation efficiency of the androgen receptor.31
Cutaneous Manifestations of Androgen Excess
Hair can be categorized as either vellus or terminal (Fig. 132-4).32 Vellus hair is fine, soft, and not pigmented; terminal hair is long, coarse, and pigmented. Virtually all hair follicles are associated with sebaceous glands to form pilosebaceous units (PSUs). The growth of the hair follicle of PSUs is controlled by as yet uncharacterized induction factor(s) elaborated by the dermal papilla. In target areas, androgens cause the prepubertal pilosebaceous unit to differentiate into either a sexual hair follicle (in which the vellus hair transforms into a terminal hair) or into a sebaceous gland (in which the sebaceous component proliferates, and the hair remains vellus). Androgens are thus necessary for sexual hair and sebaceous-gland development. Antiandrogens reverse this process, causing pilosebaceous units to revert toward the prepubertal state. Male-pattern sexual hair development (for example, mustache and beard) occurs in sites where relatively high levels of androgen are necessary for pilosebaceous-unit differentiation. The greater density of terminal hairs in the androgen-sensitive areas of men than in women is accounted for by a greater proportion of pilosebaceous units with terminal rather than vellus hairs. Male-pattern baldness is largely the result of conversion of terminal hair to sebaceous follicles.
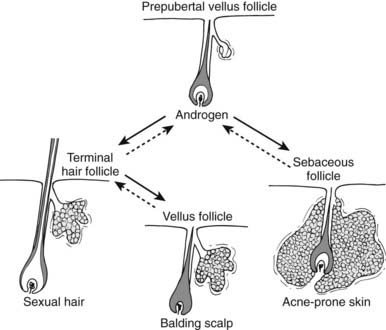
FIGURE 132-4. Role of androgen in the development of the pilosebaceous unit. Solid lines indicate effects of androgens, dotted lines those of antiandrogens. Hairs are depicted only in the anagen (growing) phase of the hair cycle. In conjunction with other regulatory factors, androgens cause the prepubertal pilosebaceous unit in androgen-dependent areas to differentiate into either a sexual hair follicle, in which the vellus hair transforms into a terminal hair, or into a sebaceous follicle, in which the sebaceous component proliferates and the hair remains vellus. In balding scalp (bottom center), terminal hairs not previously dependent upon androgen regress to vellus hairs under its influence.
(Reproduced from Rosenfield RL, Deplewski D: Role of androgens in the developmental biology of the pilosebaceous unit, Am J Med 98:80S–8S, 1995.)
Hirsutism is defined as excessive male-pattern hair growth in women.32,33 Hirsutism must be distinguished from hypertrichosis, the term used to describe the excessive growth of androgen-independent hair which is vellus, prominent in nonsexual areas, and most commonly familial or due to metabolic disorders (e.g., thyroid disturbances, anorexia nervosa) or medications (e.g., phenytoin, minoxidil, cyclosporine). In adult Caucasian and Black women, hirsutism can be defined by a total score of 8 or more on a modified scale of Ferriman and Gallwey (Fig. 132-5).34 Thus it is normal for women to have a few sexual hairs in most “male” areas (such as the upper lip and chin areas). The cutaneous manifestations of androgen excess include acne and balding in addition to hirsutism.32 However, these are variably expressed manifestations of hyperandrogenism: some patients have hirsutism or acne alone, others both, yet others neither, and balding in a diffuse pattern can be the sole manifestation.
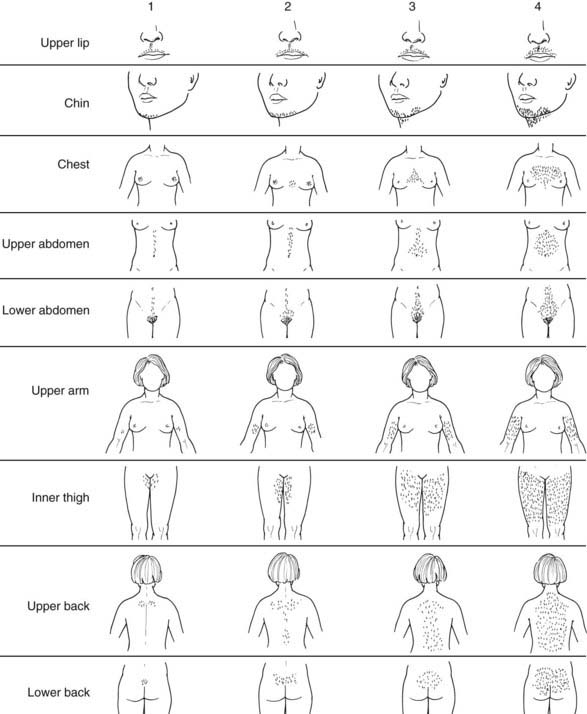
FIGURE 132-5. Hirsutism scoring scale of Ferriman and Gallwey. The nine body areas possessing androgen sensitive PSUs are graded from 0 (no terminal hair) to 4 (frankly virile) and the sum totaled. Normal hirsutism score is less than 8.
(Modified from Hatch R, Rosenfield R, Kim M, Tredway D: Hirsutism: implications, etiology, and management, Am J Obstet Gynecol 140:815–830, 1981.)
An increase in the plasma total or free testosterone concentration underlies half of the cases of mild hirsutism, and most women with a twofold or greater elevation of androgen levels have some degree of hirsutism.33 Moderately severe hirsutism or cystic acne is even more likely to be hyperandrogenic.33,35 However, it must be noted that the amount of sexual hair and sebaceous-gland development seems to depend as much upon the factors determining sensitivity of the PSUs to androgens as upon the plasma androgen level itself. At one end of the normal spectrum are women whose PSUs seem “hypersensitive” to normal blood free-androgen levels; this seems to account for “idiopathic” hirsutism and acne. At the other end of the spectrum are women whose PSUs are relatively insensitive to androgen; this seems to account for “cryptic” hyperandrogenemia (hyperandrogenism without skin manifestations).
Hyperandrogenic Disorders
Hyperandrogenism arises from abnormal adrenal or ovarian function in most cases, but occasionally from apparent abnormalities in the peripheral formation of androgen. Functional abnormalities (Table 132-3) are much more common than tumors. Polycystic ovary syndrome (PCOS) is the cause of about 80% of hyperandrogenism. Idiopathic hirsutism and idiopathic hyperandrogenemia accounts for 5% to 20%, nonclassical congenital adrenal hyperplasia for 2% to 4%, and androgen-secreting neoplasms for 0.2%.36,37 Cushing’s syndrome, acromegaly and hyperprolactinemia are rare endocrine causes of hyperandrogenism.
Table 132-3. Causes of Androgen Excess in Women
| Adrenal Hyperandrogenism |
| Gonadal Hyperandrogenism |
| Peripheral Androgen Overproduction |
ADRENAL DISORDERS
Premature Adrenarche
Normally, the first manifestation of the adrenal or gonadal contributions to androgen production is pubarche, the appearance of pubic hair.6,38 Premature pubarche refers to the appearance of sexual pubic hairs before 8 years of age in girls and is usually the result of premature adrenarche.39,40 That is, some children with the precocious onset of pubic hair have undergone a pubertal amount of maturation of adrenal androgen secretion earlier than normal. In others, premature pubarche represents inordinate sensitivity of sexual hair follicles to normal adrenal androgen levels. The best hormonal indicator of adrenarche is a plasma level of DHEAS over 40 µg/dL. In children with premature adrenarche, plasma testosterone levels also rise into the early pubertal range, and the responses of steroid intermediates to ACTH are typically in between the prepubertal and adult range. The predominant steroids are the Δ5-3β-steroids, 17-hydroxypregnenolone, and DHEA. Premature adrenarche may be a moderate risk factor for the subsequent development of polycystic ovary syndrome during adolescence. This seems particularly true in those patients who have “exaggerated adrenarche,” which is characterized by clinical features that suggest atypical severity of androgen excess and androgen levels and steroid intermediate responses to ACTH greater than those of early-pubertal girls.
The distinction of premature adrenarche from mild (nonclassical) virilizing congenital adrenal hyperplasia (CAH) has been a matter of controversy. In some clinics, as many as 40% of children presenting with premature pubarche have nonclassical CAH due to 21-hydroxylase deficiency.41,42 This high prevalence is clearly skewed by the presence of patients of ethnic groups with a high prevalence of CAH, such as Ashkenazi Jews and Hispanics. However, much of the dispute centered around the distinction of mild 3β-HSD deficiency from premature adrenarche based on the responses of steroid intermediates to ACTH stimulation. However, molecular genetic studies have subsequently shown that 3β-HSD deficiency is rarely the cause of premature pubarche and is only found when Δ5-steroid responses to ACTH are over 5 SD above normal.43 Table 132-4 shows the typical ranges of steroid intermediate responses to ACTH in normal and prematurely adrenarchal subjects.

Functional Adrenal Hyperandrogenism
Functional adrenal hyperandrogenism (FAH) may be defined as glucocorticoid-suppressible, ACTH-dependent, 17-ketosteroid excess. So defined, FAH is found in approximately a third of hyperandrogenic females in the absence of ovarian hyperandrogenism and in half of PCOS patients (see following).5,15 This entity has been attributed by some to “exaggerated adrenarche,” based upon the observation that fully half of women with 17-ketosteroid hyperresponsiveness to ACTH have a pattern of steroid intermediates which resembles an exaggeration of the steroid secretion pattern of adrenarche. Typically there are moderately excessive 17-hydroxypregnenolone and DHEA responses to ACTH, together with androstenedione hyperresponsiveness. Occasionally there are mild abnormalities of 17-OHP or 11-deoxycortisol in response to ACTH.
Currently, several lines of evidence support the concept that FAH is due to dysregulation of adrenal steroidogenesis by the same pathogenic mechanism that causes dysregulation of ovarian steroidogenesis in PCOS.44 These are: (1) The 17-ketosteroid hyperresponsiveness to ACTH can be interpreted as overactivity of 17-hydroxylase/17,20-lyase in the adrenal cortex and is frequently associated with apparent overactivity of these steps in the ovaries. (2) The adrenal overproduction of 17-ketosteroids is inconsistently associated (20% to 50%) with increased levels of the adrenarche marker, DHEAS. (3) There is evidence of widespread but variable dysregulation of other aspects of corticosteroid secretion and metabolism. In addition, PCOS brothers have elevated levels of DHEAS which correlate to the levels in the PCOS probands.45 In lean women with PCOS, a functional polymorphism of type 1-11β-hydroxysteroid dehydrogenase, which reduces the efficiency of the peripheral conversion of cortisone to cortisol, is associated with elevated DHEAS levels and increased cortisol response to ACTH.46 In addition, evidence is accumulating that hyperinsulinemia is as related to adrenal dysregulation as it is to ovarian dysregulation.
Congenital Adrenal Hyperplasia
CAH results from an autosomal recessive defect in the activity of any one of the steroidogenic enzymes necessary for the synthesis of corticosteroid hormones by the adrenal gland (see Fig. 132-1).47,48 Because of the enzyme defect, the adrenal gland cannot efficiently secrete glucocorticoids and/or mineralocorticoids. There is diminished negative-feedback inhibition of ACTH and/or of renin-angiotensin secretion, which markedly increases the secretion of these trophic hormones and leads to compensatory hyperplasia of the adrenal cortex. The result is defective synthesis of end product and accumulation of steroid precursors proximal to the enzyme defect. These precursors are subsequently converted to androgen.
Each enzyme deficiency leads to characteristic clinical syndromes and biochemical abnormalities. Virilizing CAH occurs as the result of 21-hydroxylase, 3β-HSD type II, or 11β-hydroxylase type I deficiency. In virilizing CAH, inefficient cortisol synthesis is associated with the accumulation of androgenic precursors that produce excessive virilization of female infants with genital ambiguity in the newborn period or hyperandrogenism later in childhood. These disorders may be associated with some degree of altered mineralocorticoid secretion, mineralocorticoid deficiency with salt wasting (in the case of 21-hydroxylase and 3β-HSD deficiency), and salt retention with hypertension (in the case of 11β-hydroxylase deficiency). CAH due to 21-hydroxylase deficiency accounts for over 90% of the cases of CAH. The classic forms have an overall prevalence in the general population of about 1:12,000.
Nonclassical (“late onset” or “attenuated”) 21-hydroxylase deficiency can present as premature pubarche or with peri- or postpubertal onset of hirsutism, acne, or amenorrhea. Such nonclassical cases account for about 2% to 3% of hirsutism in the general population.49,50 Some cases are entirely cryptic; they are discovered serendipitously in family studies and have no clinical manifestations. Utilizing ACTH response patterns and molecular genetic techniques, New and her colleagues51 have shown that the rapid 17-OHP response to an intravenous bolus of ACTH distinguishes among nonclassical cases and heterozygotes, and normals better than baseline steroid levels. The degree of clinical severity is correlated with the degree of 21-hydroxylase deficiency elucidated by the ACTH test.
These nonclassical forms of 21-hydroxylase deficiency CAH have been shown to be due to distinct 21-hydroxylase mutations that cause only mild enzyme deficiencies.51 Studies of the molecular biology of 21-hydroxylase deficiency have provided important tools for the diagnosis of this condition. The gene for 21-hydroxylase (P450c21) is located on chromosome 6, and DNA probes are now available to detect the most common mutations.
Severe 3β-HSD deficiency leads to defective cortisol and mineralocorticoid synthesis, with adrenal insufficiency presenting in the newborn period. Because of deficient synthesis of gonadal Δ4-steroids (including testosterone), affected male infants classically have insufficient virilization, with sexual ambiguity. Affected female infants, on the sother hand, are often virilized because of the very high levels of the weakly androgenic DHEA and its more androgenic metabolites.52 The controversy about distinguishing mild 3β-HSD deficiency from simple premature adrenarche has been discussed earlier under functional adrenal hyperandrogenism.
11β-hydroxylase deficiency classically presents with genital ambiguity in the female and subsequent incomplete sexual precocity in association with hypertension. Atypical cases with late-onset hirsutism without hypertension have been reported but are very rare.53
Adrenal Neoplasms
Adrenal tumors are a very rare cause of hyperandrogenism. Only two adrenal tumors were found in three series of hyperandrogenic women totaling over 2100 patients.36,37,54 The combination of onset of symptoms distinct from menarche, rapid progression of symptoms, signs of virilization, and elevated androgen levels usually make the diagnosis of tumor straightforward. Only about half of patients with virilizing adrenal tumors will have a marked elevation of DHEAS (>700 µg/dL).55 If an androgen-secreting tumor is suspected, pelvic ultrasound should be done. If negative, it should be followed by an adrenal CT scan.
Other Adrenal Disorders
Glucocorticoid-suppressible hyperandrogenism may also occur in the setting of hyperprolactinemia and acromegaly. The symptoms of hyperprolactinemia may mimic those of PCOS: hirsutism or acne, menstrual disturbance, and galactorrhea are typical, although not necessarily present.56 Polycystic ovaries and LH hyperresponsiveness to GnRH may be associated with a prolactinoma.57,58 DHEA and DHEAS levels are elevated, as is the plasma free testosterone, in part because of a direct effect of hyperprolactinemia on adrenal cortical function. Mild to modest elevations in prolactin levels may be found in otherwise classic PCOS. In these instances, it is important to establish that the prolactin elevation is not a consequence of primary hypothyroidism, prolactinoma, or medications. Acromegaly may also be associated with either adrenal or ovarian androgenic hyperfunction, possibly because of direct effect of the elevated IGF-1.59
A number of other causes of adrenal hyperandrogenism may occur, but presentation due to hirsutism or menstrual disturbance is relatively rare. These include Cushing’s syndrome,60,61 glucocorticoid resistance,62 or excessively rapid cortisol metabolism.63
OVARIAN DISORDERS
Most women with androgen excess have ovarian hyperandrogenism, as demonstrated by the results of ovarian vein catheterization,64 dexamethasone suppression testing,34,64–66 long-term administration of GnRH agonists,67,68 and more recently, the acute ovarian steroidogenic response to the administration of a GnRH agonist66,69 or human chorionic gonadotropin (hCG).70 The causes of gonadal androgen excess are listed in Table 132-3.
Polycystic Ovary Syndrome (PCOS)
PCOS is the most common endocrine disorder in reproductive-aged women, with a prevalence between 5% and 10%.71–73 Variance in reported prevalence may reflect the impact of ethnic origin, race, and environmental factors that can modify the expression of the PCOS phenotype.74,75
Seventy-five years after its first description by Stein and Leventhal,76 diagnostic criteria for PCOS have evolved considerably. It is now generally accepted that PCOS can be diagnosed as a disorder of otherwise unexplained hyperandrogenic anovulation (National Institute of Health criteria).77 Because the ovarian dysfunction of PCOS may not be reflected in anovulatory symptoms, and infertility is relative rather than absolute, the alternative diagnostic “Rotterdam criterion” of a polycystic ovary (PCO) with evidence of hyperandrogenism was proposed78 and is widely accepted.49 Although the Rotterdam criteria recognize much of the clinical heterogeneity of the syndrome, they are not without controversy. They include two groups of patients who do not fulfill the National Institutes of Health criteria: (1) patients with polycystic ovaries who are hyperandrogenic but ovulate normally, and (2) patients with anovulatory dysfunction and polycystic ovaries but no evidence of hyperandrogenism. The Androgen Excess Society has endorsed the first of these groups, considering a polycystic ovary to constitute alternative evidence of ovarian dysfunction. However, they dispute the second of these criteria, arguing that patients who have no evidence of hyperandrogenism should not be given a diagnosis of PCOS.49
Clinical Characteristics
The symptoms of PCOS usually begin at about the time of menarche,79 but postpubertal onset is also seen, usually as a result of environmental modifiers such as excess weight gain. Anovulatory symptoms (oligo/amenorrhea, dysfunctional uterine bleeding, and/or infertility) occur in about 75%, and most of the rest may have subclinical anovulation with apparent eumenorrhea.49,80 Hirsutism affects about 60%, acne 15% to 20%, and androgenic alopecia about 5% of PCOS patients. Obesity has been reported in 30% to 75% of PCOS subjects, the majority of whom have an android distribution of body fat with a disproportionate amount of visceral adipose tissue. An android body-fat distribution is also often present in PCOS subjects of normal weight.81
The definition of PCO, when evaluated by transvaginal ultrasonography, is that at least one ovary demonstrates an ovarian volume greater than 10 cm3 (10.5 cc if volume computed by formula for prolate ellipsoid) or 12 or more follicles measuring 2 to 9 mm in diameter. In adolescents, the criteria are volume greater than 10.8 cm3 and 10 or more follicles per maximum plane.82 A PCO is found in approximately 75% of women with PCOS.49,83,84 However, about 20% of women who report regular menses and have no hirsutism also have PCO; they seem to have a slightly lower ovulation rate than women with normal ovaries.85 About half of women with a PCO have normal ovarian function, but about half have subclinical ovarian dysfunction that is related to PCOS, with half in turn of the latter group meeting Rotterdam criteria for PCOS, albeit without obvious clinical manifestations.82
About 60% to 80% of PCOS patients will have elevated levels of androgens. It is unclear why hyperandrogenemia is not always demonstrable; it is likely that much of this has to do with inadequate methodology.33 The most sensitive indicator of hyperandrogenemia is the plasma free-testosterone level. DHEAS levels are elevated in about 25% of PCOS, and in 10% it may be the sole abnormality in circulating androgens.49
The vast majority of PCOS patients have functional ovarian hyperandrogenism (FOH).15 Typically (in approximately 70%) they have an elevated 17-OHP response to GnRH agonist–induced endogenous gonadotropin release.66,82 In vivo and in vitro challenges with LH or the LH analog, hCG, result in markedly elevated 17-OHP responses and, to a lesser extent, elevated androgens in PCOS patients or their theca cells, compared with controls.86,87 There is no evidence of a steroidogenic block. In addition, plasma free testosterone and 17-OHP remain abnormally high after adrenocortical function is suppressed by dexamethasone. Dexamethasone suppression and GnRH agonist test results are 85 percent concordant in PCOS.66 Those PCOS subjects who lack 17-OHP hyperresponsiveness to gonadotropin stimulation (approximately 30% of cases) have a unique type of ovarian dysfunction that is relatively hyposensitive to gonadotropin.88
Adrenal hyperandrogenism is also common in PCOS. As noted above, about 25% will have elevated DHEAS levels. Approximately half of patients with PCOS also have a characteristic adrenal dysfunction which coexists with FOH or occurs independently of it in about equal numbers. This primary FAH is characterized by moderately excessive responses of dehydroepiandrosterone and 17-hydroxypregnenolone to ACTH, without evidence of a block in steroidogenesis. This has been postulated to represent the adrenal manifestation of a steroidogenic dysregulation characteristic of PCOS.89
Excessive LH secretion relative to FSH was the first laboratory abnormality identified in PCOS.90 Hypersecretion of LH occurs in about 90% of PCOS subjects, as evidenced by an exaggerated LH response to GnRH, elevated mean LH levels, increased LH pulse amplitude and increased LH pulse frequency.83 The most obese subjects are the least likely to have LH elevation.83 The LH secretory abnormalities in PCOS are more likely a result of anovulation and ovarian hyperandrogenism than its cause. Progesterone inhibits pulsatile LH secretion, and there is no cyclic production of progesterone in PCOS. LH levels and pulse frequency are less sensitive than normal to suppression by exogenous estradiol and progesterone, although they fall sufficiently for androgen levels to be normalized.91,92 Antiandrogen treatment of adult PCOS normalizes the inhibitory effect of progesterone and estradiol on LH pulse frequency.93 These results suggest that androgen excess in PCOS interferes with the inhibitory feedback of estradiol and progesterone resulting in LH hypersecretion.
Metabolic Abnormalities
Both lean and obese women with PCOS are generally more insulin resistant than normal women matched for total and fat-free body mass.81 About 65% to 70% of women with PCOS are insulin resistant.94 It has long been recognized that women with PCOS are at increased risk for developing type 2 diabetes.95 Impaired glucose tolerance (IGT) or frank diabetes is evident in approximately 45% of women with PCOS by their fourth decade.96,97 Long-term follow-up of women with PCOS substantiates an increased prevalence of type 2 diabetes, compared with that of appropriate control groups. The conversion rates from normal glucose tolerance to either IGT or type 2 diabetes are also high in women with PCOS.81
The synergistic effects of insulin resistance and obesity were thought to be sufficient to account for the predisposition to diabetes in PCOS. However, elevated glucose levels are evidence that the ability of the pancreatic β cell to compensate for insulin resistance has been exceeded. Alterations in insulin secretion are demonstrable in PCOS, and these alterations may have a genetic basis, since they are present more often in those women with PCOS who have a first-degree relative with type 2 diabetes.81,98
Nonalcoholic fatty liver disease is associated with insulin resistance. Ultrasound evidence of fatty liver occurs significantly more frequently in obese PCOS patients than in obese controls and is associated with hyperinsulinemia and insulin resistance. Nonalcoholic fatty liver disease was present in 41% of obese PCOS subjects in one series, and half of those had elevations of ALT, suggesting the possibility of the more severe nonalcoholic steatohepatitis.99
Dyslipidemia is reported in up to 70% of PCOS patients. Various patterns have been described, the most common one being increased levels of LDL cholesterol, VLDL cholesterol, and triglycerides, and reduced levels of HDL cholesterol (particularly decreased HDL-2, the most antiatherogenic HDL subtype), compared with control women. Qualitative disorders of LDL cholesterol have also been described, characterized by an increased amount of small, dense LDL cholesterol, which is an independent predictor of cardiovascular morbidity and mortality.81
Women with PCOS do not consistently demonstrate an increased risk of hypertension, but they may be at increased risk for its development later in life. Obesity may be the major influence on the risk of hypertension in PCOS.81
Given the above findings, it is not surprising that women with PCOS are at increased risk for metabolic syndrome.81 Up to 43% of nondiabetic women with PCOS meet diagnostic criteria for metabolic syndrome well before the end of their fourth decade, and most of these before the end of their third decade of life. Indeed, the prevalence of metabolic syndrome in young PCOS women is similar to that in women between the ages of 50 and 60 years. The prevalence of metabolic syndrome is similar across racial backgrounds. Obesity and possibly ethnicity do, however, contribute to the development of metabolic syndrome. For instance, the prevalence of metabolic syndrome was lower among a less obese group of Italian women with PCOS than among a more obese group of American women with PCOS.100
There is evidence of early coronary and other vascular disease in women with PCOS. Compared with age-matched controls, women with PCOS have increased intima media thickness in carotid arteries, a condition linked to cardiovascular risk factors that include dyslipidemia and obesity and also an independent predictor of stroke and myocardial infarction. Another marker of atherosclerosis, coronary artery calcification, is more common in women with PCOS than in controls, even after adjusting for age and BMI. Echocardiography has revealed anatomic and functional differences between women with PCOS and controls. Women with PCOS have an increased left atrial size and left ventricular mass index, and a reduced left ventricular ejection fraction. Of note, the left ventricular mass index is linearly related to the degree of insulin resistance. Diastolic dysfunction has also been noted to disproportionately affect women with PCOS.81
Given the above risk factors, are women with PCOS at increased risk for coronary heart disease? The largest study of long-term consequences of PCOS reported to date is by Wild and Pierpoint and colleagues. They determined risk factors and cause of death in a cohort of 786 PCOS women diagnosed between 1930 and 1979 in the United Kingdom. Standardized mortality ratios (SMR) were used to compare the causes of death in the PCOS cohort to national rates. PCOS women had a higher prevalence of risk factors for cardiovascular disease, including diabetes, hypertension, and high cholesterol. The PCOS subjects had a significantly increased risk of death from diabetes (SMR 4.6) but not of death from all causes (SMR 0.93) or cardiovascular disease (SMR 0.78). There was an increased risk of endometrial cancer (odds ratio 6.1) and cerebrovascular disease (odds ratio 3.4) in the PCOS subjects and a nonsignificantly increased risk of coronary heart disease (odds ratio 1.2).101,102
The Nurses’ Health Study found a significantly increased risk of coronary heart disease in women who had a history of irregular and very irregular menstrual cycles (a reasonable surrogate for the presence of PCOS). The relative risk was 1.53 in women with very irregular periods, which is within the confidence intervals of the risk reported for coronary heart disease in the study by Wild et al. The association was stronger in overweight (BMI ≥ 25) than in lean women. The risk was unchanged when women with menopause before the age of 40 were excluded from the analysis.103
Pathogenesis
The pathogenesis of PCOS has been heavily debated, probably a result of the fact that more than one pathogenic mechanism can result in the endpoint of a polycystic, hyperandrogenic ovary. Increasing evidence suggests that PCOS arises as a complex trait with contributions from both heritable and environmental factors.39,104 About 50% of sisters of PCOS probands have hyperandrogenemia, and about half of those will also be anovulatory.105 There is evidence that the PCO of PCOS is inherited as an autosomal dominant trait, with mothers typically being asymptomatic.106,107 Diabetes-related defects in insulin secretion and action are common in primary relatives of women with PCOS.108–110 In a Dutch twin family study, the correlation between monozygotic twins for PCOS was 0.71, and for dizygotic twin or non-twin sister pairs the correlation was 0.38.111 These family studies demonstrate a significant genetic component in the pathogenesis of PCOS. Investigators have studied the contribution of more than 70 genes to the PCOS phenotype, and reasonable evidence for a role in PCOS have been found for a few.104 Association studies have found that variants in CYP11A alleles, which encode p450 side chain–cleavage enzyme, may play a role in the etiology of PCOS. There is an association of the A8 allele of the fibrillin-3 gene (FBN3) with PCOS and with hyperinsulinemia and insulin resistance in PCOS.104,112 There is an inverse relationship between androgen receptor activity and the number of CAG repeats encoding polyglutamine in the N-terminal transactivation domain of the androgen receptor (AR). Because the AR gene is X-linked, the pattern of X-inactivation could influence AR activity and the PCOS phenotype. Sisters with the same AR CAG repeat genotype and the same phenotype (PCOS or normal) more frequently show the same pattern of X-inactivation than do sisters with discordant phenotypes.104 Well-studied candidate genes which do not have a role in PCOS include the insulin gene, VNTR, and CAPN10.104
In some cases, the intrauterine environment may affect the pathogenesis of PCOS. PCOS is common in females with congenital virilizing disorders.39 CAH is the most frequent, but not exclusive, cause of this outcome. The diagnosis of PCOS is established in CAH by finding elevation of testosterone when anovulatory symptoms persist in women in whom the adrenal dysfunction of CAH is well controlled on glucocorticoid therapy. It seems to be present in about half of women with classic CAH but occurs in only perhaps a fifth of women with nonclassical CAH.113,114 The PCOS of CAH is associated with a higher degree of LH hyperresponsiveness to GnRH-agonist testing than found in ordinary PCOS. Rhesus monkeys exposed to androgen excess early in gestation will develop classic PCOS features115,116; these animals have ovarian and adrenal hyperandrogenism, oligomenorrhea, polyfollicular ovaries, and elevated LH levels. They also have abdominal obesity, insulin resistance, impaired glucose tolerance, and dyslipidemia. Analogous to classic PCOS, the LH hypersecretion seen in congenitally virilized cases of PCOS appears to result from congenital androgen exposure interfering with the negative-feedback effect of ovarian steroids on LH.39
Low birth weight for gestational age (LBW) may predispose to PCOS in adolescence and adulthood. There is a well-established association between intrauterine growth restriction and postnatal insulin resistance,117 which makes an association between LBW and PCOS plausible. In addition, LBW is associated with early adrenarche in most populations studied.39 Ibañez and colleagues have reported that among Catalan girls with exaggerated adrenarche, postmenarcheal follow-up found that 52% were not hyperandrogenic, 25% had PCOS without hyperinsulinemia, and 23% had both PCOS and hyperinsulinemia. The birth weights of these successive groups averaged 0.25, 1.0, and 2.0 SD below the mean.118 Thus, increasing degrees of intrauterine growth restriction seemed to be associated with increasing severity of disease from premature adrenarche to PCOS, and finally to PCOS and hyperinsulinemia. However, follow-up of birth cohorts in British, Finnish, and French populations found no relationship of LBW and PCOS.39 In the British cohort, it was heavy babies whose mothers’ weights were above average who developed PCOS.
Ovarian hyperandrogenism accounts for most, if not all, of PCOS. When functional adrenal and functional ovarian hyperandrogenism coexist, it is most like due to common underlying pathogenic mechanisms. Early in vitro studies using minced ovarian tissue, adrenal and ovarian vein catheterization studies, and adrenal and ovarian suppression studies showed that the ovary was the primary androgen source in PCOS.119 We found that women with classic PCOS have a characteristic pituitary/ovarian response to testing with an acute GnRH-agonist challenge test.66,69,120 Their gonadotropin responses resemble those of males in that they have significantly greater early LH and lesser FSH responses than normal females. They have greater 17-OHP and, to a lesser extent, androgenic responses than those of normal women. 17-Hydroxypregenenolone and DHEA responses are less consistently elevated. There is no evidence of a discrete block in the steroidogenic pathway, since the responses of all steroids on the pathway from 17-OHP to estrone and estradiol are normal or elevated. The elevated responses of 17-OHP and androstenedione, without evidence of a block in the subsequent steroidogenic steps or consistent abnormalities in the earlier steps in the steroidogenic pathway, suggest increased ovarian 17-hydroxylase and 17,20-lyase activities as part of a generalized overactivity of steroidogenesis. This dysregulation of ovarian steroidogenesis can manifest in the absence of LH excess or sonographic abnormalities.
Dysregulation of ovarian androgen secretion appears to result from escape from desensitization to LH and not from excessive LH secretion.5 This may be the consequence of abnormal modulation of LH responses by endocrine, paracrine, or autocrine factors which normally coordinate intraovarian androgen with estrogen production.15 Normal theca cells are very sensitive to the down-regulating effect of LH levels within the physiologic range. Maximal stimulation of 17-OHP and androstenedione secretion by theca cells in culture normally occurs at LH concentrations approximating the upper portion of the normal range for follicular phase serum LH levels, and a further increase in LH dosage leads to no further rise. However, both in vivo and in vitro studies suggest that the steroid responses of PCOS do not fall along the normal LH steroid dose-response curve, but rather seem displaced upwards and to the left. Thus, the defect in steroidogenesis appears to be the result of escape from normal down-regulation of theca-cell secretion, rather than from overstimulation by LH.
The cause of the escape of theca cells from down-regulation could be intrinsic or extrinsic to the ovary. IGFs and inhibins are candidates for being FSH-inducible ovarian factors capable of interfering with down-regulation of ovarian steroidogenesis.15 Insulin is a key endocrine factor because it amplifies the effect of gonadotropins on steroidogenesis.5 The abnormality of thecal function in PCOS resembles the escape from desensitization which occurs when normal theca cells are treated with insulin or IGF.
Studies of theca-cell cultures support an intrinsic thecal abnormality in PCOS. In primary culture, PCOS theca cells secrete excessive amounts of androgens and androgen precursors, suggesting a generalized up-regulation of steroidogenic enzymes.86 Long-term cultures of passaged PCOS theca cells secrete increased amounts of progesterone, DHEA, 17-OHP, androstenedione, and testosterone per cell, both basally and after cAMP stimulation, in comparison to theca cells from women who are normally ovulatory.3,121 This is due to the overexpression of mRNA for most steroidogenic enzymes. These findings persist when insulin is absent from the culture medium. Thus, the persistent increased androgen production of the PCOS ovary is a result of up-regulation of specific steroidogenic enzymes and is consistent with an intrinsic theca-cell abnormality rather than an effect of the in vivo paracrine and endocrine milieu. This concept is supported by the finding of persistent hypersensitivity to gonadotropin after long-term gonadotropin suppression in typical PCOS.88
The pathogenesis of PCOS may also involve primary abnormalities of folliculogenesis. The polycystic ovary accumulates increased numbers of 2 to 8 mm, small antral follicles. These follicles are no more likely to be atretic than follicles of similar size in normal ovaries, and they produce more estradiol in response to FSH. Some of these small antral follicles are prematurely responsive to LH, which implies the acquisition of LH receptors, which only occurs in the dominant follicle in the normal ovary.122 More recent studies have confirmed that there are increased numbers of primary follicles in the anovulatory, polycystic ovary.122 There is also a higher percentage of follicles that have initiated growth and increased granulosa-cell proliferation in preantral PCOS follicles. The anovulation of PCOS is more likely a result of these abnormalities of folliculogenesis than of androgen-induced follicular atresia.122
Since so little is known about the regulation of preantral folliculogenesis in the normal ovary, the cause of abnormal early folliculogenesis in the PCOS ovary is far from clear. But some progress has been made. Anti-müllerian hormone (AMH), which suppresses preantral follicle growth in mice, is decreased in early, growing PCOS follicles. Thus, the increased numbers of small antral follicles in the PCOS ovary may be mediated in part by a lack of AMH.122 In the rhesus monkey, testosterone implants increase the number of both healthy preantral and small antral follicles.123 Prenatal exposure to androgens is associated with aberrant folliculogenesis in both the sheep and rhesus monkey.122 While it is possible that a primary theca-cell abnormality may result in abnormal early folliculogenesis, evidence of abnormal folliculogenesis exists after long-term suppression of ovarian androgen excess, which suggests that this defect is fundamental to the disorder.88
Insulin resistance and hyperinsulinemia also play an important role in the pathogenesis of PCOS. Insulin synergizes with LH to stimulate theca-cell steroid production to a much greater degree in PCOS than in normal ovaries,124,125 and treatment with insulin-sensitizing agents reduces androgen levels in PCOS.126,127 Insulin appears to act via its own receptor in theca cells by a signaling pathway separate from that mediating its metabolic effects.125 Insulin also inhibits liver production of sex hormone–binding globulin, which may increase testosterone bioavailability.
Distinct postreceptor binding defects in insulin signaling have been described in fibroblasts, adipocytes, and skeletal muscle cells in PCOS.81,128 These defects are specifically associated with impaired glycogen synthesis and glucose uptake and do not involve mitogenic signaling. Interestingly, glucose metabolism is normal after long-term culture of adipocytes and skeletal muscle cells, despite persistence of the signaling defects in the muscle cells.128
Ovarian hyperandrogenism and insulin resistance/hyperinsulinemia have become the new “vicious cycle” of PCOS. It is clear that each can aggravate the other, but which of the two is primary and which secondary is controversial. Women with extreme insulin resistance due to insulin receptor antibodies or to genetic abnormalities of the insulin receptor or postreceptor pathways have severe ovarian hyperandrogenism and virilization.39,124 In rare instances, severe insulin resistance can be the primary pathogenic event in PCOS. However, insulin resistance can result from primary hyperandrogenic conditions. Untreated women with nonclassical CAH have insulin resistance similar to that seen in PCOS.129,130 In addition, testosterone, acting through its own receptor, induces resistance to the metabolic effects of insulin in cultured adipocytes from normal women.131 Treatment with the antiandrogen, flutamide, for 12 months significantly reduces visceral fat mass and improves insulin sensitivity.132
At this point in time, the majority of evidence suggests that the primary abnormality in PCOS is one of theca-cell androgen production rather than insulin resistance. Visceral fat has increased lipolytic activity, and free fatty acid levels are increased in PCOS.133 In turn, free fatty acids cause insulin resistance in skeletal muscle.134 Thus it has been speculated that hyperandrogenemia may affect adipocyte insulin sensitivity in such a way as to favor visceral body-fat distribution, which results in relative increases of free fatty acids, which induce skeletal muscle insulin resistance.135 An alternative hypothesis has recently emerged from the observation that an insulin-responsive transcription factor involved in fat and glucose metabolism up-regulates the testosterone-forming gene (17β-HSD type V).136 This raises the possibility that the multiple defects of PCOS arise from dysregulation of transcription factors that are common to multiple pathways of metabolism in multiple tissues.
Attempts have been made to find a common pathogenic mechanism for insulin resistance and ovarian and adrenal hyperandrogenism in PCOS. The postreceptor binding defect in PCOS fibroblast insulin receptors is characterized by increased serine phosphorylation, which inhibits the intrinsic tyrosine kinase activity of the insulin receptor and is a result of a serine kinase extrinsic to the receptor.137 Increased serine phosphorylation of P450c17 increases its 17,20-lyase activity, which could result in increased androgen production by both the ovary and adrenal in PCOS. Thus a gain-of-function mutation of a serine kinase targeting the insulin receptor and P450c17 could result in both the insulin resistance and hyperandrogenism of PCOS.138 Unfortunately there is no experimental evidence supporting this hypothesis.139
PCOS may also arise in the setting of frankly virilizing disorders and ovarian steroidogenic blocks. Poorly controlled virilizing CAH or exogenous androgen administration to transsexual females have been shown to produce the ovarian anatomic changes of Stein-Leventhal syndrome, namely cortical sclerosis, cysts, and hyperthecosis, probably because of the great increase in the intraovarian concentration of androgen.5 The virilization of classic CAH can also cause FOH. Women with virilizing CAH may have polycystic ovaries and high serum LH levels.140 Adrenal rests of the ovaries, as are found in rare cases of CAH, may also mimic PCOS.113,141 Ovarian steroidogenic blocks in the pathway to estradiol may on rare occasions cause hyperandrogenism, estrogen deficiency, and multicystic ovaries. This can result from congenital defects in the activity of ovarian 3β-HSD,69 17β-HSD,142 or aromatase143,144 (see Fig. 132-1). The aromatase mutations reported to date have resulted in congenital virilization with ambiguous genitalia.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


