FIGURE 111-1. Mature forms of human atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). Bold circles represent amino acids common to the three human hormones. The biologically active forms shown are ANP, BNP-32, and CNP-22.
BIOCHEMISTRY
Synthesis and Structure
In humans, the two genes that encode ANP and BNP are arranged in tandem (approximately 8 kb apart) on the short arm of chromosome 1. Transcription of the ANP gene yields a 151 amino acid precursor, preproANP, which is cleaved to produce the 126 amino acid prohormone (proANP), the main storage form and the chief constituent of atrial granules.3 Final processing of stored proANP by the cardiac membrane serine protease (corin)4 takes place during the hormone’s release into the circulation and culminates in the formation of ANP99-126 (or ANP-28), the major bioactive and circulating form (see Fig. 111-1) and the amino-terminal (NT) fragment ANP1-98. Constitutive secretion of ANP-28 and proANP may also occur from the ventricle in pathologic states such as left ventricular hypertrophy and heart failure. BNP gene transcription and processing in humans yields a 134 amino acid precursor, followed by a 108 amino acid propeptide, which is cleaved by an unknown protease, yielding the mature 32 amino acid BNP77-108 (or BNP-32) (see Fig. 111-1) and the bio-inactive NT fragment BNP1-76.2 In the ventricle, BNP appears to be predominantly secreted directly from myocytes via a constitutive pathway. Both forms circulate in human plasma, together with a higher molecular weight component, probably the prohormone itself (proBNP1-108).
Distinct from ANP and BNP, the gene that encodes CNP is located on chromosome 2 in humans and is transcribed to produce a 126 amino acid precursor molecule that yields a 103 amino acid proCNP peptide. Processing of proCNP appears to be specifically mediated intracellularly by the enzyme furin to yield an NT fragment (NTproCNP1-50) and CNP-53 (proCNP51-103), which is the predominant biologically active form of the peptide in tissues such as the vascular endothelium.2 CNP-53 is processed further at unknown extracellular sites to yield CNP-22 (proCNP82-103), the mature form of the hormone found in the circulation that retains full biological activity. In contrast to NTproCNP,5 plasma CNP-22 levels are low and are close to the limits of detection in normal adults.
Factors that increase ANP and BNP gene expression are closely linked to myocyte hypertrophy and are highly conserved across species and in different pathologic states. Members of the GATA family of transcription factors play a dominant role.6 As with ANP, BNP transcripts and secretion are markedly increased in states of chronic left ventricular strain and overload. Both angiotensin II (AngII)7 and hypoxia-inducible transcription factor (HIF)8 increase BNP gene expression in rat cardiomyocytes. Less is known about transcriptional regulation of CNP gene expression, although the Wnt signaling pathway and the transforming growth factor (TGF)-β–stimulated transcription factor, TSC-22, have been identified as positive regulators.2
Tissue Distribution
The major site of ANP gene expression is the cardiac atrium, which in health produces approximately 95% of total secreted ANP. The normal ventricle also expresses the gene but at a level 100-fold lower than in the atrium. However, expression in the ventricle may increase markedly in pathologic states such as congestive heart failure and ventricular hypertrophy, and may become an important source of circulating forms of ANP.3 Extracardiac ANP gene expression occurs (although at much lower levels) in the lung, aortic arch, adrenal medulla, gastrointestinal tract, immune system, eye, and central nervous system. In the kidney, processing at amino acid 94/95 yields urodilatin (proANP95-126), which may have unique intrarenal paracrine functions.2 As with ANP, the highest levels of BNP are found in atrial tissue (albeit at 5% of ANP levels), where it is co-stored with ANP within a subset of atrial granules. Concentrations of ventricular BNP mRNA are similar to those of atrial tissue. Other sites of BNP synthesis include the brain (except in humans), kidney, lung, thyroid, spleen, and amnionic tissue. CNP was first described in the central nervous system, where the concentration of the peptide exceeds that of either ANP or BNP, suggesting a functional role for CNP as a neuropeptide. CNP and transcripts have also been identified outside the central nervous system, most notably within vascular endothelial tissue, where the peptide may serve as a vasodilator and inhibitor of vascular cell proliferation, and the pituitary. Low levels of CNP have also been found in cardiac fibroblasts9 and in a range of other tissues, including kidney, blood cells, cornea, skeletal tissue, and reproductive organs. Enormously high concentrations of CNP are reported in seminal fluid plasma, presumably sourced from seminal vesicles.10
Receptors and Metabolism
To date, three different subtypes of natriuretic peptide receptor (NPR) have been identified.2 Two of these subtypes, NPR-A and NPR-B, are coupled to particulate guanylate cyclase (GC) and appear to mediate most of the well-known actions of the natriuretic peptides through production of the intracellular second messenger, cyclic guanosine monophosphate (cGMP). Splice variants of the NPR-B, including a species lacking a functional GC domain, have also been identified, but whether these variants participate in signaling is unknown.2 NPR-A selectively binds both ANP and BNP (with preference for ANP) and is present in high concentration in the kidney (particularly within the glomerulus and the inner medullary collecting duct), adrenals, heart, vascular smooth muscle, lung, adipose tissue, and brain. NPR-B is highly specific for CNP,2 and its distribution overlaps to some extent that of NPR-A, although it is also found in reproductive tissue and the skeleton. The third (and most prevalent) receptor subtype, NPR-C, binding all three natriuretic peptides with high affinity, has no GC domain and is thought to function largely as a “clearance” receptor by internalizing the peptide for lysosomal degradation.2 However, receptor stimulation has been shown to inhibit adenyl cyclase in several tissues (including vascular smooth muscle cells [VSMCs]), to stimulate phosphoinositide hydrolysis, and to mediate some of the antimitogenic effects of the natriuretic peptides.11 The physiologic significance of these in vitro findings remains unclear. NPR-C is widely distributed and appears to represent most of the natriuretic peptide receptors in vascular endothelial and smooth muscle cells; it is also prominent in the heart, adrenal gland, kidney, and skeleton. In humans, the affinity of NPR-C for BNP is appreciably less than for ANP and CNP.2
In addition to regulation by transcription factors, the functional activity of natriuretic peptide receptors is affected by activation, desensitization, and receptor cross-talk in vivo. Both NPR-A and NPR-B genes are regulated by several Sp1 binding sites within the promoter region. Activity of both receptors is reduced by chronic exposure to ligand (homologous desensitization)—actions that appear to be mediated in part by cGMP. Activation of both NPR-A and NPR-B involves change in the phosphorylation state; similarly dephosphorylation is a mechanism of receptor desensitization.2 Of note, in rodents there appears to be differential reduction in NPR-A activity in the failing heart, whereas NPR-B activity is increased.12 Known inhibitors of NPR-A/NPR-B include the mitogens sphingosine-1-phosphate (S-1-P), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and vasoconstrictors such as AngII, arginine vasopressin (AVP), and ET, all of which activate protein kinase C (PKC) and raise intracellular ionic calcium levels. These findings provide a molecular basis for the largely inhibitory effects of growth factors (e.g., AngII, bFGF, PDGF, ET) and vasoconstrictors (AngII, AVP, and ET) on the vasodilator and antiproliferative actions of natriuretic peptides.2
Receptor-mediated endocytosis of natriuretic peptides via NPR-C is an important degradative pathway that affects the local concentration of synthesized hormone. A recently identified endogenous NPR-C ligand, osteocrin,13 presumably has similar effects. The other major pathway involved in metabolism of the natriuretic peptides is enzymatic cleavage by neutral endopeptidase (NEP) 24.11 (neprilysin). This enzyme, a membrane-bound zinc metalloproteinase that exhibits broad substrate specificity, is present in many epithelial tissues, including the vasculature, lung, and kidney, where it is concentrated in the proximal tubular membrane. A soluble form of the enzyme is also detectable in human plasma. In humans and mice, BNP is much more resistant to enzyme hydrolysis than is ANP or CNP. However, after hydolysis by the renal enzyme meprin A yielding BNP7-32, the truncated peptide loses this resistance—at least in mice.14 Together with its lower affinity for NPR-C, these differences may in part explain the prolonged plasma half-life of BNP (approximately 22 minutes) in humans compared with ANP and CNP (2 to 4 minutes). Nevertheless, the metabolic clearance rates of all three hormones are similar in adult humans (approximately 2 to 4 L/min), suggesting a markedly greater volume of distribution of BNP than of the other two peptides.
CONTROL MECHANISMS
The most important stimulus to the secretion of ANP is an increase in atrial wall tension.3 Despite the generally strong relationship between atrial pressure and ANP secretion, discrepancies can occur, as has been observed during cardiac tamponade and after acute intravenous volume loading. In addition to atrial stretch, many other factors, including mechanical factors such as heart rate (particularly the frequency of atrial contraction) and numerous physiologic and pathophysiologic factors (e.g., increased sodium intake, supine posture, water immersion, exercise, hypoxia, myocardial ischemia, hypertrophy), may affect secretion of the peptide. In addition, multiple chemical stimuli (particularly ET and AngII but also adrenergic agonists, acetylcholine, glucocorticoids, calcium ionophores, prostaglandin F2α, vasopressin, calcitonin gene-related peptide [CGRP], growth factors, thrombin, platelet-activating factor, relaxin, corticotropin-releasing factor, estradiol, and ANP itself) may directly stimulate ANP release or enhance its response to acute volume load.3 Conversely, nitric oxide (NO), testosterone, and possibly AM inhibit ANP secretion.
Responses of BNP are similar to those of ANP in many circumstances. Like ANP, plasma BNP is increased by chronic (dietary) sodium loading and other hypervolemic states (e.g., chronic renal failure, some forms of hypertension). In most of these situations, the increases in plasma ANP and BNP are comparable; however, BNP appears to be less responsive than ANP to abrupt changes in intracardiac pressure (e.g., acute changes in posture, exercise, rapid ventricular pacing, acute saline loading). These findings presumably reflect the relative content of the two hormones within the atrial granules. The primary stimulus to BNP secretion is distending pressure within the ventricles (i.e., hemodynamic [wall] stress). Proportionately greater increases in plasma BNP (vs. ANP) concentrations occur after acute myocardial infarction,2 in patients with heart failure, and in hypertrophic cardiomyopathy. These latter differences are consistent with the markedly augmented gene expression of BNP compared with ANP in overloaded ventricles and contrast with the relatively unchanged levels of atrial BNP mRNA and the secretion observed in these states.
Although little is currently known about the regulation of CNP in vivo, studies directed toward identifying a vascular CNP system15 provide evidence that a variety of factors affecting vascular tone and cell growth have the capacity to stimulate CNP production in vitro. Transforming growth factor (TGF)-β, interleukin-1β, tumor necrosis factor (TNF)-β, thrombin, lipopolysaccharide, bFGF, and endotoxin (most of which are derived from VSMCs, macrophages, and platelets) have been shown to enhance CNP secretion from cultured endothelial cells. Shear stress and oxidative stress also increase CNP synthesis in endothelial cells. On the other hand, CNP secretion is inhibited by oxidized low-density lipoprotein, insulin, and vascular endothelial cell growth factor. With the use of human VSMCs, serum and PDGF-BB both increase CNP gene expression.16 CNP secretion from cardiac fibroblasts is stimulated by TGF-β, bFGF, and ET.9 In the heart, CNP concentration in ventricular myocardium is reported to increase threefold in severe heart failure, and similar increases in CNP gene expression occur in rodent cardiac ventricle after myocardial infarction. These findings are consistent with a small (positive) venoarterial CNP gradient across the heart in humans17 and in experimental animals.18
Outside the cardiovascular system, CNP gene expression in the adult murine uterus is stimulated by estrogen, fluctuates in phase with the estrus cycle in rats, and changes profoundly in uterine and placental tissues during gestation.19 Except for TGF-β (which stimulates CNP gene expression in osteoblasts and chondrocytes), regulation of CNP in skeletal tissues is poorly understood despite its crucial role in endochondral bone growth.20
FUNCTION
The natriuretic peptides exhibit a broad range of effects in tissues subserving body fluid homeostasis, as well as effects that maintain the structural integrity of cardiovascular tissue (Fig. 111-2). Apparently standing outside these actions is the recently recognized role of the natriuretic peptides (especially CNP) in endochondral bone formation.
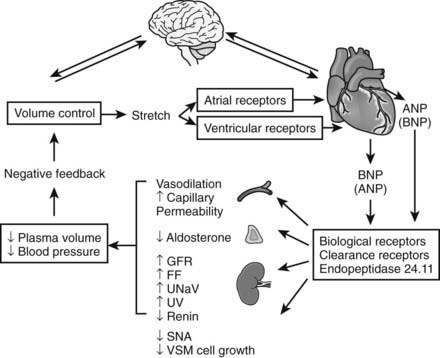
FIGURE 111-2. Schematic diagram showing the regulation and integrated actions of atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP). Possible regulation of cardiac synthesis and secretion of ANP/BNP by the brain and actions of circulating natriuretic peptides on circumventricular brain tissues are discussed in the text. Auto/paracrine actions of the natriuretic peptides (including C-type) in cardiovascular and other tissues are omitted for reasons of clarity. FF, Filtration fraction; GFR, glomerular filtration rate; SNA, sympathetic nervous activity; UNaV, urine sodium excretion; UV, urine volume; VSM, vascular smooth muscle.
(From Rademaker MT, Espiner EA: The endocrine heart. In Becker KL [ed]: Principles and practice of endocrinology and metabolism, ed 3, Philadelphia, 2001, Lippincott Williams & Wilkins, pp 1622–1634).
Many of the systemic actions of natriuretic peptides, particularly in the kidney, are influenced by a variety of other physiologic variables, including resting arterial blood pressure, sodium status, the degree of neurohormonal activation, and age. It is intriguing to note (with only a few exceptions) that the physiologic effects of the natriuretic peptides appear to be counterpoised with those of the RAAS (see Receptors/Metabolism). Functional outcome therefore is determined in part by receptor status and by opposing or synergizing intracellular mediators, in addition to the local concentration of the bioactive form of the hormone. Physiologic responses are elicited by the intracellular production of cGMP and its downstream effector protein kinases (cGMP-dependent protein kinases), which in VSMCs reduce intracellular ionic calcium concentration.2
Renal Effects
Administration of ANP in normovolemic states induces prompt natriuresis and diuresis (mediated by NPR-A) and smaller increases in divalent ions without significantly elevating potassium excretion. Larger doses of ANP have been shown to elevate the glomerular filtration rate (GFR). The most striking renal effect of ANP is that it increases the fractional excretion of sodium (the fraction of total sodium filtered at the glomerulus that is excreted in the urine), acting largely at tubular sites. ANP has also been shown to redistribute blood flow to the deeper nephrons with less sodium reabsorptive capacity. A number of the proposed actions of ANP appear to be mediated by inhibition of the action of AngII and, to a lesser extent, AVP and aldosterone. It seems likely that natriuretic potency and other renal actions of BNP are closely similar to those of ANP in humans.21 In contrast, CNP lacks natriuretic activity, even when administered at high doses directly into the renal artery, and its role in renal pathophysiology remains unclear.
As was noted earlier, many of these actions of ANP and BNP are attenuated in pathophysiologic states such as chronic heart failure (CHF). The precise reasons are still the subject of debate but could include downregulation of NPR-A by high circulating concentrations of ligand, increased intrarenal renin-AngII/ET, impaired processing of prohormones, and a disproportionate fall in circulating bioactive forms.22
Hemodynamic Effects
One of the best documented hemodynamic effects of ANP was a fall in arterial (chiefly systolic) blood pressure (see Fig. 111-2), which in normal health is mediated largely by a fall in cardiac output consequent to reduced cardiac filling pressure. Some of this effect can be attributed to a reduction in plasma volume (as evidenced by an increase in venous hematocrit) associated with a shift of plasma from the vascular to the extravascular space. This latter action (see Fig. 111-2) is mediated by enhanced microvascular permeability of the endothelium.23 ANP shortens atrial conduction time and effective refractory period in atria of intact human hearts. However, cardiac muscle contractility is largely unaffected by either ANP or BNP, whereas coronary blood flow is increased and myocardial oxygen consumption is decreased. A further important hemodynamic action of ANP is to reduce sympathetic tone in the peripheral vasculature. These actions are mediated by a direct inhibitory effect on sympathetic nerve outflow, in addition to sensitizing of vagal afferents with consequential reflex bradycardia and dampening of arterial baroreflex responses.24 Other actions contributing to the hypotensive effect include early natriuresis/diuresis, inhibition of renin and aldosterone secretion, and inhibition of the vasopressor response to AngII and ET. Although the hemodynamic effects of BNP in humans are similar to those of ANP,21 venodilation appears to be a more prominent action than arterial relaxation—at least at physiologic concentrations of the hormone. This suggests that a major action of BNP is to reduce cardiac preload rather than afterload.
A range of studies have shown that CNP has vasodilator activity that is enhanced by removal of the endothelium. Proportionately greater venodilator effects (mediated by NPR-B) have been observed in contrast to the possible arterial bias of ANP.25 In normotensive humans, high-dose infusions of CNP reduced systemic blood pressure without altering heart rate or cardiac output (suggesting a reduction in peripheral vascular resistance), whereas lower, more physiologic doses had no hemodynamic effect.25 CNP has also been shown to induce vasorelaxation of coronary arteries and the pulmonary vasculature. Earlier reports that CNP acted as an endothelially derived hyperpolarizing factor (EDHF) have not been confirmed.26
Endocrine Effects
The antagonism of AngII by natriuretic peptides extends to many endocrine actions (e.g., inhibition of AngII-induced aldosterone and vasopressin secretion) and other central effects mediated by the brain RAAS. In humans, inhibition of renin secretion appears to be a consistent and prompt effect of ANP (and BNP) administration; a direct inhibitory effect on renin release by ANP is supported by in vitro studies. It is also likely that inhibition is mediated by increased delivery of sodium and chloride to the macula densa. ANP and BNP have a selective inhibitory effect on the zona glomerulosa, whereby aldosterone production is reduced (mainly via inhibition of cholesterol uptake into mitochondria). In contrast to the cardiac natriuretic peptides, antagonism of the RAAS by CNP appears negligible.
Antiproliferative Effects
All three natriuretic peptides (ANP, BNP, and CNP) have antiproliferative (antimitogenic) effects in a variety of tissues, including vascular smooth muscle, adrenal gland, kidney, brain, myocardiocytes, and endothelial cells.2 In the heart, ANP inhibits hypertrophy of cultured myocardiocytes and impairs left ventricular modeling after cardiac injury.27 ANP and BNP, acting via NPR-A, inhibit ET-1 and AngII-stimulated 3H-thymidine incorporation into cardiac fibroblasts. Further in vitro evidence of the antifibrotic effects of BNP is provided by studies showing that the hormone inhibits the profibrotic actions of TGF-β in primary human cardiac fibroblasts.28 Inhibiting effects of both hormones on cardiac fibrosis may also be mediated by actions reducing the activity of matrix metalloproteinases.2 CNP, previously considered to have little or no role in cardiac tissue, potently inhibits cardiac fibroblast proliferaton in vitro,9 as well as cardiomyocyte hypertrophy induced by ET-1.25 Consistent with these in vitro findings are reports of reduced cardiac hypertrophy and preserved contractility in rats with experimental myocardial infarction when given infusions of CNP. Taken together, these studies suggest important roles for all three peptides in cardiac remodeling—findings vindicated by studies using genetic modification and highlighting the importance of the autocrine/paracrine actions of natriuretic peptides as cardioprotectors (see also below).
CNP also has well-established antiproliferative and antihypertrophic actions in VSMCs, presumably mediated by NPR-B. CNP reduces neointimal formation and thrombosis induced by injury to the arterial wall. These anti-inflammatory effects of CNP appear to be mediated at least in part by NO generation.29 On a related theme, CNP acting via NO and cGMP pathways reportedly enhances angiogenesis and neovascularization in an in vivo model of hind limb ischemia.30 Similar observations of CNP-induced capillary density have been reported in the myocardium of rodents with experimental autoimmune myocarditis.31 How CNP mediates endothelial regeneration is unclear, but the hormone may promote, rather than initiate, angiogenesis at a specific phase as mature blood vessels are being formed.31
Neuroendocrine and Other Effects
Expression of all three natriuretic peptides (particularly CNP) and their receptors in discrete and diverse regions of the brain and spinal cord32 suggests a central (brain) natriuretic peptide system that is analogous to the brain RAAS. Even though ANP and BNP do not cross the blood-brain barrier, the presence of NPR-A in subfornical and circumventricular tissues suggests that these circulating hormones may affect the central neural tissues that regulate blood pressure and fluid homeostasis (see Fig. 111-2). Mechanisms whereby natriuretic peptides affect neurotransmission and their functional consequences are still largely unknown, but studies with ANP and CNP showed actions on norepinephrine and dopamine release in some cell preparations, including cells in the spinal cord, where changes were associated with increases in cGMP. Of note, neuroprotection (prolonging neuronal survival) has been attributed to a natriuretic peptide–induced increase in cGMP concentrations, for example, in settings of metabolic and hypoxic stress.32 CNP appears to act as a classical growth factor regulating proliferation, patterning, and axonal pathfinding in brain33 and spinal cord.34
Contrary to their antiproliferative activities, all three natriuretic peptides (particularly CNP) have been shown to stimulate the growth of osteoblasts in vitro. CNP appears to be the most potent of any of the known growth factors (on a molar basis) in stimulating fetal tibial elongation and chondrogenesis.35 This apparent paradox may be explained by the reciprocal antagonism between CNP cell signaling and that of FGF,36 which in skeletal tissue is antimitogenic37 as opposed to mitogenic in other tissues. As discussed below, the importance of CNP in endochondral growth—by expanding the proliferative and especially the hypertrophic zone of growth plates—is now well established.20 Other emerging roles for the natriuretic peptides, for example, in reproductive and placental tissues (CNP),19 in adipocytes (ANP),2 and in immune function (ANP),38 continue to unfold.
ROLE IN PHYSIOLOGY AND PATHOPHYSIOLOGY
The striking range of actions of the natriuretic peptides (e.g., reductions in cardiac filling pressure, arterial vasodilation, promotion of sodium and water excretion, inhibition of the RAAS, reduced baroreflex sensitivity) provides strong circumstantial evidence that they play a fundamental role in maintaining blood pressure and volume. More concrete evidence is supplied by studies showing that blockade of the natriuretic peptide system increases plasma renin-aldosterone, ET, and norepinephrine levels; reduces sodium excretion and GFR; and accelerates the development of hypertension and heart failure. However, it has been the advent, over the past decade, of techniques employing genetic modifications in rodents that has revealed the important autocrine and paracrine actions of natriuretic peptides, in addition to confirming the role of ANP and BNP as circulating hormones.6 Phenotypes of mice resulting from gene manipulations affecting ANP/BNP (Table 111-1) or CNP (Table 111-2) highlight the mostly distinct actions of these hormonal systems mediated by NPR-A and NPR-B, respectively, with overlap as expected in studies in which the NPR-C gene is disrupted. Collectively, these studies expose the important antifibrotic and antihypertrophic actions of cardiac tissue production of ANP and BNP—effects that are independent of hemodynamic load, as shown by experiments using NPR-A disruption targeted to the ventricle. In addition, these studies have illustrated the importance of effective processing of the prohormone,4 the importance of normal circulating ANP concentration and activity for regulation of blood pressure and sodium excretion,6 and, for the first time, pinpointed the critical role of the vascular endothelium in effecting acute and chronic actions of ANP on blood pressure and plasma volume.23 Unexpectedly, these studies also revealed a new role for natriuretic peptides in bone biology,6,20 leading to the unmasking of the part played by the CNP signaling pathway in endochondral growth (see Table 111-2). Despite in vitro evidence of the role of CNP in the microvasculature,2 no cardiovascular phenotype was obvious when the CNP pathway was blocked or augmented. However, transgenic rats overexpressing a dominant negative mutant of NPR-B (effectively reducing the number of functional receptors), while still dwarfed, developed cardiac hypertrophy.25 These results suggest that paracrine actions of cardiac CNP secretion are indeed cardioprotective. This view is further supported by studies showing decreased cardiac hypertrophy after experimental myocardial infarction in mice in which the CNP gene was overexpressed in cardiomyocytes.39
Table 111-1. Phenotypes of Genetic Modifications Affecting ANP/BNP
| Gene Modification | Effect |
|---|---|
| ANP overexpression | Hypotension |
| BNP overexpression | Hypotension; skeletal overgrowth |
| ANP knockout | Hypertension; cardiac hypertrophy |
| BNP knockout | Cardiac fibrosis |
| Corin knockout | Hypertension; high plasma proANP, low ANP |
| NPR-A overexpression | Hypotension |
| NPR-A knockout | Hypertension; cardiac hypertrophy |
| NPR-A–targeted knockout | |
| Heart muscle | Cardiac hypertrophy |
| Vascular smooth muscle | Loss of acute hypotensive action of ANP |
| Vascular endothelium | Plasma volume expansion; hypertension |
| NPR-C knockout | Hypotension; hypovolemia; skeletal overgrowth |
ANP, Atrial natriuretic peptide; BNP, B-type natriuretic peptide; NPR, natriuretic peptide receptor.
Table 111-2. Phenotypes of Genetic Modifications Affecting CNP
| Gene Modification | Effect |
|---|---|
| CNP knockout | Dwarfism; early death |
| CNP overexpression (targeted to chondrocytes) | Skeletal overgrowth |
| CNP overexpression (targeted to heart muscle) | Prevents cardiac hypertrophy post MI |
| NPR-B knockout | Dwarfism; uterine and ovarian hypoplasia |
| NPR-B dominant negative (overexpression) | Dwarfism; cardiac hypertrophy |
| NPR-C knockout | Skeletal overgrowth; hypotension and hypovolemia |
CNP, C-type natriuretic peptide; MI, myocardial infarction; NPR, natriuretic peptide receptor.
In humans, plasma venous levels of immunoreactive ANP in young healthy adults range from 5 to 25 pmol/L and show little evidence of pulsatile secretion or diurnal rhythmicity. Concentrations increase with age and are elevated by supine posture, high dietary sodium intake, and volume expansion. Circulating BNP levels in normal humans are consistently lower (0.3 to 10 pmol/L) than those of ANP.2 Recent work shows that a variety of BNP immunoreactive species, including BNP 3-32 (the product of dipepidyl peptidase IV cleavage), proBNP1-108, and possibly glycosylated forms, circulate in human plasma. Of these, only the 3-32 analogue and mature BNP 1-32 are bioactive,22 and the presence of the latter in human (systemic) circulation has even been questioned. These findings need to be borne in mind when one interprets concentrations measured by different techniques. Although it is still unexplained, BNP levels vary inversely with body mass index, plasma insulin, and triglycerides.40 Both plasma ANP and BNP tend to be higher in females and increase during estrogen therapy. BNP is also affected by thyroid status independent of cardiac dysfunction, and is positively correlated with increases in thyroid hormone values. Similar to ANP, BNP levels are higher in the elderly and increase in sodium-loaded states, but show less fluctuation than ANP with acute maneuvers such as changes in posture and exercise. ANP-BNP interactions have been noted, whereby the addition of either one increases the steady state plasma concentration of the other, possibly through dissociation of pre(receptor)-bound hormone.41 As was previously mentioned, plasma ANP and BNP levels are elevated in a number of hypervolemic states, in keeping with increased intracardiac pressure. The most striking increases occur in congestive heart failure, where both ANP and BNP concentrations correlate with severity.2 These marked increases in plasma levels are largely caused by augmented ventricular secretion of both peptides. Other disorders of cardiac muscle (e.g., dilated cardiomyopathy) or valvular function are also associated with elevated circulating levels of cardiac natriuretic peptides. Plasma ANP and BNP are increased soon after myocardial infarction, even in the absence of clinical cardiac decompensation, with concentrations reflecting infarct size and the increase in cardiac filling pressure. Both ANP and BNP are increased after subarachnoid hemorrhage (BNP more than ANP), possibly as a consequence of transient subendocardial ischemia associated with intense cardiac sympathetic nervous activation. Increased concentrations of ANP and BNP in chronic renal failure also may reflect “volume overload” inasmuch as levels fall after dialysis (ANP more than BNP). Raised levels of both hormones are observed in pulmonary hypertension (reflecting right ventricular dysfunction)2 in patients with acute lung injury and in those with a variety of other noncardiac edematous states, including the nephrotic syndrome and cirrhosis,2 in which central blood volume may be increased. Notably, BNP is markedly elevated in critical illness, especially severe sepsis,42 yet correlates poorly with pulmonary capillary wedge pressure in patients under intensive care.43 Significant, but smaller, increases in both ANP and BNP levels occur in hypertension and correlate with left ventricular hypertrophy. A variety of tachyarrhythmias may markedly increase plasma hormone levels (ANP more than BNP in humans).
CNP is present at very low plasma concentrations in healthy adults (0.4 to 3 pmol/L), whereas the aminoterminal fragment (NTproCNP) is some 10- to 50-fold higher and is readily measurable.5 These differences presumably reflect their relative half-life (2 to 3 minutes for CNP) in plasma. CNP values, especially NTproCNP, are increased in actively growing children in keeping with increased growth plate activity and endochondral bone growth. Transorgan regional studies report a small but highly significant positive venoarterial gradient for both CNP and NTproCNP across the heart in subjects being investigated for coronary artery disease. Circulating levels of both CNP and NTproCNP increase in subjects with heart failure, although the rise is small compared with ANP and BNP. Elevated plasma CNP levels (4 to 36 pmol/L) have been reported in patients with severe sepsis, chronic renal failure (with levels decreasing significantly after hemodialysis), cor pulmonale, hypoxia, and aneurysmal subarachnoid hemorrhage. Whether these increases constitute a response to specific stimuli or reflect widespread endothelial damage or impaired clearance is unknown. Of interest, and consistent with findings on the role of CNP in skeletal growth, elevated plasma forms of CNP occur in subjects with CNP overexpression caused by chromosomal rearrangements, and in children with hormonal resistance due to loss of function mutations in NPR-B.20
To date, few validated reports have described a primary disorder of natriuretic peptide secretion or action resulting in disease. Exceptions are homozygous loss of function in NPR-B, causing extreme short stature (AMDM, acromesomelic dysplasia Maroteau type20), and overexpression of CNP associated with skeletal overgrowth, kyphosis, and other complications. As yet no cardiovascular phenotype has been reported in these subjects. Polymorphisms and deleterious mutations in genes coding for ANP or BNP, or components of the signaling pathway, have been reported in association with hypertension, left ventricular hypertrophy, myocardial infarction, and other vasculopathies,44 but for obvious reasons are more difficult to detect than disorders of skeletal growth. However, a recent report of familial atrial fibrillation associated with a heterozygous frame shift mutation in the ANP gene, together with increased circulating levels of a mutant ANP form,45 is an important finding and strongly suggests that many other significant mutations affecting the cardiovascular system in humans will emerge.
CLINICAL APPLICATIONS
Diagnosis/Prognosis
The close relationship between natriuretic peptide plasma levels and cardiac load has led to their use in clinical diagnosis and as markers of cardiac dysfunction. Raised plasma concentrations of ANP and, in particular, BNP and its N-terminal fragment (NTproBNP) not only serve as markers of left ventricular dysfunction but also predict subsequent hemodynamic deterioration in asymptomatic subjects and the development of overt heart failure.2 Early reports46 that plasma BNP concentration could be used to distinguish heart failure from other causes of acute dyspnea have been confirmed,47 and it is this indication that is now seen as the most useful clinical application of BNP/NTproBNP measurements.48 A low concentration of the hormone is particularly useful in excluding decompensated heart failure in the setting of acute dyspnea. A large and increasing body of evidence indicates that measurement of BNP/NTproBNP can assist in the evaluation of a variety of disorders affecting the cardiovascular system, some of which are summarized in Table 111-3. NTproBNP and BNP assays appear to be equally useful and are now recommended in current guidelines for the management of patients with heart failure.49 Risk management of cardiovascular disorders using measurements of BNP forms is likely to be improved by the addition of other markers of myocardial injury such as troponin I.
Table 111-3. Clinical Uses of BNP/NTproBNP Measurement
BNP, B-type natriuretic peptide.
Treatment
Although circulating concentrations of both ANP and BNP are markedly elevated in decompensated heart failure, the possibility that administration of pharmacologic doses of these or related hormones may improve outcome has prompted their use in selected patients. Following reports of beneficial effects of long-term administration of ANP in acute heart failure, the U.S. Food and Drug Administration in 2002 approved the use of recombinant BNP (nesiritide) in this setting. Nesiritide infusion was associated with increased natriuresis, decreased plasma aldosterone and endothelin, and significant hemodynamic and symptomatic improvement in selected patients.2 Subsequent and wider use has raised concerns related to adverse effects, including hypotension and impaired renal function, that are still to be laid to rest. Other drugs such as ularitude (urodilatin) are being evaluated, although numbers of patients treated to date are small.
The therapeutic potential of ANP as treatment for peripheral vascular disease, refractory to other treatments, has recently been tested.50 When it was administered continuously by IV infusion for 2 weeks, symptoms (including intermittent claudication and rest pain) were relieved and healing of foot ulcers was promoted. Improvement was attributed both to vasodilation and possibly to augmented angiogenesis.
In view of the marked antiproliferative actions of natriuretic peptides within the heart itself, interest in the concept of cardioprotection using appropriate peptide agonists in the setting of cardiac injury, for example, after myocardial infarction, has been renewed. ANP and BNP agonists carry risks of unwelcome hypotension and renal impairment that are less likely when CNP agonists are used. Moreover, the latter may be as effective—or more so—in preventing left ventricular remodeling. Evidence that NPR-B may account for most natriuretic peptide activity in the rodent failing heart12 is another reason to explore new approaches using drugs that target both NPR-A and NPR-B receptors. One such “chimeric” drug, CD-NP,51 demonstrates renal actions consistent with NPR-A activation, is less hypotensive when compared with BNP in dogs, and, consistent with the incorporation of CNP-22 within the molecular structure, is more potent than BNP in stimulating cGMP from human cardiac fibroblasts. Results from studies in humans using this or similar compounds are awaited with interest.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


