FIGURE 5-1. Schematic of G protein–coupled receptor (GPCR) structures. Top panel represents a subclass I rhodopsin/neurotransmitter GPCR. Middle panel represents a subclass II glucagon–related receptor. Bottom panel represents a subclass III metabotropic glutamate–related receptor.
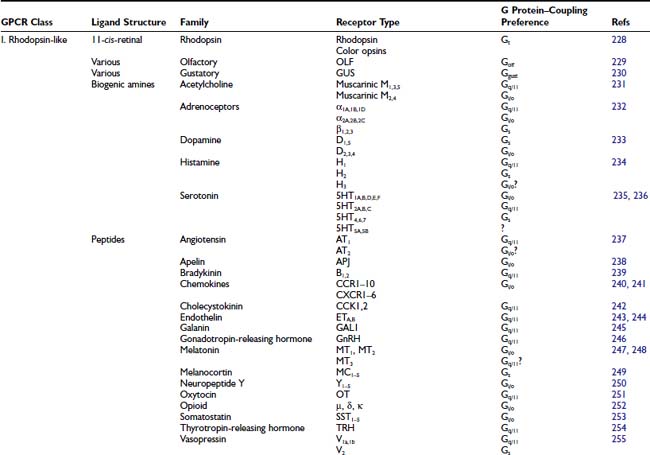
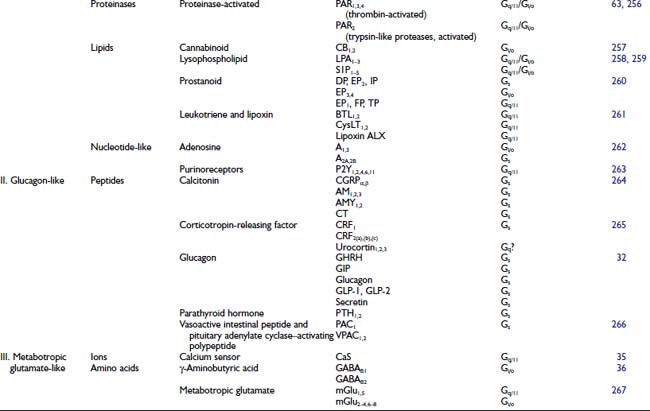
Subclass I rhodopsin-like receptors, which form the largest GPCR subgroup, include receptors that have a wide diversity of agonists, including light, neurotransmitter, and glycoprotein hormones.27 Subclass I contains receptors for 31 agonist families (see Table 5-1), with each family including as many as 13 members (serotonin receptors). The separate receptors all are encoded on different genes. Functionally distinct isoforms of the dopamine D2 receptor are generated by alternative exon splicing.28
The overall sequence homology among subclass I receptors is low, being restricted to several highly conserved amino acids located for the most part in the cytoplasmic half of the transmembrane core (see Fig. 5-1). Mutagenesis experiments suggest that many of these highly conserved amino acids contribute to protein stability and to the conformational changes that mediate receptor activation.29–31 The only residue that is conserved among all subclass I receptors is an arginine in the Asp-Arg-Tyr motif at the cytoplasmic side of transmembrane helix (TM) 3. Two cysteine residues in the second and third extracellular loops, which are conserved in most GPCRs, form a disulfide bridge that has been implicated in the packing and stabilization of the transmembrane bundle (see Fig. 5-1).
The glucagon-related receptors (subclass II) include a relatively small group of peptide receptors that are expressed in endocrine cells of the pancreas and the gastrointestinal epithelium, and in specialized neurons in the brain (see Table 5-1).32 Glucagon, glucagon-like peptide-1, glucagon-like peptide-2, glucose-dependent insulinotropic peptide, growth hormone–releasing hormone, and secretin are structurally related peptides that exert their actions through secretin-related class II receptors.32 Except for the disulfide bridge between the second and third extracellular loops, subclass II receptors do not contain any of the conserved structural features that characterize subclass I receptors such as the Asp-Arg-Tyr motif. These receptors share a relatively large aminoterminus extracellular domain (≈100 residues) that contains several cysteines that form a network of disulfide bridges.
The metabotropic glutamate-related receptors (subclass III) are characterized by an extremely long aminoterminus extracellular domain (≈500 to 600 residues) that is implicated in ligand binding.33 This subclass includes the metabotropic glutamate receptors (mGluRs),34 the calcium sensing receptors,35 the γ-aminobutyric acid (GABAB) receptor,36 the vomeronasal mammalian pheromone receptors,37 and the putative taste receptors.38 Except for a disulfide bridge between extracellular loops 2 and 3, subclass III receptors do not share any conserved residues with subclasses I and II.
Structural Features of G Protein–Coupled Receptors
GPCRs have an extracellular aminoterminus, seven α-helical transmembrane spans (which form the transmembrane core), three extracellular loops, three intracellular loops, and an intracellular carboxyterminus (see Fig. 5-1). Each of the seven transmembrane spans generally is composed of 20 to 27 amino acids. In different GPCRs, the terminus (7 to 595 amino acids), loops (5 to 230 amino acids), and carboxyterminus (12 to 359 amino acids) vary considerably in length.
Conserved GPCR sequences are largely contained within the hydrophobic transmembrane domains. To facilitate comparison of corresponding residues among different Class I receptors, several numbering schemes have been developed. The Schwartz and Baldwin numbering schemes are similar.39,40 In these schemes, the most conserved residues in each helix are numbered according to their predicted relative position in a standard helix of 26 amino acids. A given residue then is described by the helix in which it is located (I through VII) followed by a number indicating its position in the helix. For example, II.9 corresponds to residue number nine in transmembrane span two. One limitation of this approach, which leads to differences between the two related systems, is that the beginning of the helix cannot be assigned unequivocally. In the Ballesteros numbering scheme,41 the most conserved amino acid in each transmembrane span is given the arbitrary number 50, and each amino acid is numbered according to its position relative to this conserved residue. For example, 4.57 indicates a residue located in transmembrane span four, seven residues toward the carboxyterminus from Trp(4.50), the most conserved amino acid in helix four. In this chapter, the residues will be indicated according to the Ballesteros numbering.41 Thus, the index residues in each of the TMs of bovine rhodopsin are Asn1.50, Asp2.50, Arg3.50, Trp4.50, Pro5.50, Pro6.50, and Pro7.50. All of these are highly conserved among rhodopsin-like GPCRs; therefore this approach allows unambiguous alignment of the transmembrane spans of these receptors.
What advantages are conferred by this widely adopted seven transmembrane template? An odd number of transmembrane spans place the aminoterminus and the carboxyterminus at opposite membrane surfaces. This allows ligand binding and receptor glycosylation at the aminoterminus, and phosphorylation and palmitoylation at the carboxyterminus (see later). We can speculate that seven transmembrane spans may be the minimum necessary to form a stable yet flexible transmembrane core with sufficient size and versatility to offer specificity, regulatory mechanisms, and contact sites for G proteins and other signaling molecules.
The first crystal structure of any GPCR, the structure of bovine rhodopsin, was solved at 2.8 Å resolution.42 The determination of the structure of rhodopsin at atomic resolution represented a milestone in the study of GPCRs and transmembrane signaling, and confirmed the existence of seven transmembrane (TM) helices (Fig. 5-2). However, rhodopsin is highly specialized for the detection of light, and it exhibits functional and biochemical characteristics that distinguish it from GPCRs for hormones and neurotransmitters. Crystal structures have recently been determined for the β2-adrenoceptor,43–45 the β1-adrenoceptor,46 native opsin—the ligand-free form of rhodopsin47—and the adenosine A2A receptor.48 The seven TM segments are arranged as a closed loop in a clockwise direction for TM1 to TM7, as viewed from the intracellular surface. A fourth intracellular loop is anchored by palmitoyl groups attached to a pair of cysteine residues, forming an eighth cytoplasmic amphiphilic helix that lies along the surface of the cell membrane. Sequence conservation suggests a similar helical structure (H8) among other subclass I GPCRs.
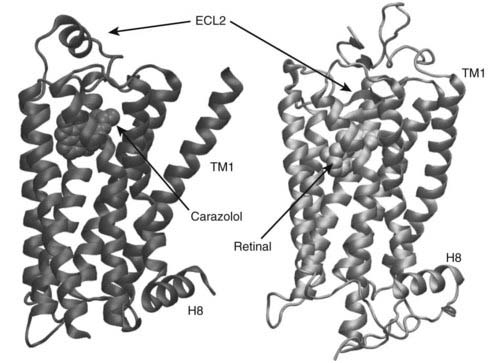
FIGURE 5-2. Comparison of rhodopsin and inverse agonist-bound β2-adrenergic receptor. Extracellular loop 2 (ECL2) of rhodopsin forms a lid over the retinal-binding pocket, whereas the position of ECL2 of the β2-adrenergic receptor allows relatively free access to the carazolol-binding pocket.
Within the inner leaflet of the plasma membrane, TM4 and TM6 are perpendicular to the lipid bilayer plane, whereas TM1, TM2, TM3, and TM5 have a lateral tilt, and TM7 is kinked inward in the center (see Fig. 5-2). In this arrangement, the core primarily comprises TM1, TM2, TM3, TM5, TM6, and TM7, whereas TM1 and TM4 are located peripherally. The inner sections of TM2 and TM3 are nearly parallel, and both helices form a nucleus for packing the other TM helices. A network of hydrogen bonds forms in the middle of the heptahelical core, thus constraining rhodopsin in the ground state. This network is mediated in part by the side chains of the highly conserved Asn(1.50)-Asp(2.50) pair in helices I and II (see Fig. 5-2) and Trp(4.50) in helix IV. The cytoplasmic side of the helical core is organized mainly by hydrophobic interactions. These interactions are arranged in two layers parallel to the lipid bilayer. One of the layers overlies the hydrogen-bond network in the middle of the heptahelical core described above, and the other surrounds the highly conserved Glu/Asp-Arg-Tyr (3.49–3.51) motif (see Fig 5-2).
Superimposition of the β2-adrenergic receptor and dark rhodopsin structures reveals substantial differences in the relative disposition of the TM helices, particularly in TMIII, IV, and V (see Fig 5-2).49–51 In rhodopsin, the second extracellular loop contains two short antiparallel β-strands that pair with two strands from the N-terminus to form a four-stranded β-sheet that buries retinal. In the β2-adrenoceptor, the N-terminal peptide is disordered, and the second extracellular loop forms an α helix that is locked in position by two disulfide bonds and a number of hydrophobic packing interactions. The position of the helix leaves the ligand-binding site accessible to the extracellular environment, as might be expected for a receptor that binds diffusible ligands. The structures of the turkey β1-adrenoceptor and the human β2-adrenoceptor overall are very similar. An interesting difference occurs in the second intracellular loop, which is thought to be involved in G protein activation. In the β1-adrenergic structure, this loop contains a short α helix, whereas the β2-adrenergic structure adopts a less regular structure.
Posttranslational Modifications
GLYCOSYLATION
In common with most membrane proteins, a majority of GPCRs have at least one glycosylation site in their N-terminal domain.52 A few GPCRs, such as the α2B-adrenoceptor, lack identifiable glycosylation sites. In glycosylated GPCRs, high-mannose, complex or hybrid oligosaccharides are linked to the Asn side chain (N-linked glycosylation) through a multistep process.
The first step of N-glycosylation involves the co-translational transfer of Glc3Man9GlcNAc2 from the lipid carrier dolichol pyrophosphate oligosaccharide onto the nascent protein by oligosaccharide transferase, a process that occurs in the lumen of the rough endoplasmic reticulum. All types of N-linked glycans share a common pentasaccharide core structure. The glycan is attached to the Asn residue in the Asn-Xxx-Thr/Ser consensus sequence. This consensus sequence must be oriented correctly and accessible for glycosylation to occur. Not all consensus sites are glycosylated.
As the glycoprotein is processed through the smooth endoplasmic reticulum and the Golgi apparatus, the oligosaccharide is trimmed and elaborated. The initial step of this processing is the removal of the three glucosyl residues, which results in a high-mannose type of chain. Complex oligosaccharides contain additions to the core glycan, which include galactosyl, fucosyl, sialyl, and GlcNAc residues.
The functional significance of glycosylation differs among individual GPCRs. The oligosaccharide moieties are important for the expression and stability of the gonadotropin-releasing hormone (GnRH) and V1a receptors but do not contribute to high-affinity agonist interaction. Likewise, glycan chains are essential for correct folding and trafficking of the vasoactive intestinal peptide (VIP)-1, the thyrotropin-releasing hormone (TRH) receptor, and the follicle-stimulating hormone (FSH) receptor. For some GPCRs, including somatostatin, β2-adrenergic, TRH, and gastrin-releasing peptide receptors, glycosylation is important for high-affinity ligand binding and may contribute to receptor G protein coupling. For many GPCRs, however, glycosylation has no known function. This latter group includes oxytocin, histamine (H2), M2 muscarinic acetylcholine, NK1, bombesin BB1, adenosine A2a, and angiotensin (AT2) receptors. Although N-linked glycosylation of GPCRs is almost universal, its influence on the properties of the mature protein is variable and unpredictable.
Palmitoylation
Covalent lipid modifications anchor numerous signaling proteins to the cytoplasmic face of the plasma membrane. These modifications mediate protein-membrane and protein-protein interactions and often are essential for function.53 Protein fatty acylation occurs through amide linkages (N-acylation) or thioester linkages (S-acylation). N-Acylation occurs on the aminoterminal glycine residue following removal of the initiator methionine by a methionyl-aminopeptidase. S-acylation occurs on cysteine residues through a thioester linkage in a wide variety of sequence contexts. Palmitate is the most commonly used S-linked fatty acid. This posttranslational process usually is referred to as protein palmitoylation. However, other fatty acids can be incorporated into cellular proteins by a thioester linkage, including myristate, stearate, and arachidonate.
Palmitoylation is a posttranslational modification that is restricted to a small subset of cellular proteins, among which proteins involved in signal transduction are prevalent. This thioesterification of cysteine residues by palmitate is distinguished from other lipid modifications by its reversibility. Indeed, in contrast to myristoyl and prenyl moieties that are added co-translationally and generally remain attached to the proteins until protein degradation, the protein-bound palmitate is added posttranslationally. Moreover, the palmitoylation state of several proteins is regulated dynamically. In particular, biologic regulation of the palmitoylation state of the G proteins and of their cognate receptors has been demonstrated.
Many GPCRs have been shown to be palmitoylated at cysteine residues in the intracellular C-terminal tail, including rhodopsin, β2– and α2-adrenoceptors, luteinizing hormone/chorionic gonadotropin, endothelin ETA and ETB, and vasopressin V2 receptors. The 5-HT1A and 5-HT1B serotonin, dopamine D1 and D2, and mGlu4 receptors also have been reported to be palmitoylated; however, the actual sites of palmitoylation for these receptors have not been demonstrated. A mutant µ-opioid receptor with its two cysteines in the carboxyterminus that was replaced was still palmitoylated, suggesting that palmitoylation of this receptor must occur at another position.
Palmitoylation serves to enhance the association of cytosolic proteins with the membrane. Palmitoylation of GPCRs anchors the C-terminal tail to the plasma membrane, creating in essence a fourth intracellular loop. Elimination of palmitoylation sites attenuates G protein coupling of β2-adrenoceptors, endothelin ETB and somatostatin SST5 receptors.53 Initial activation of the β2-adrenoceptor promotes rapid depalmitoylation of both the receptor and the Gα protein subunit, and sustained activation prevents palmitoylation from occurring. Palmitoylation has been found to be obligatory for ligand-promoted ERK/MAPK activation by the endothelin ET receptor.54 However, for many GPCRs, palmitoylation is not essential for receptor–G protein coupling.55
The palmitoylation state of the receptor governs internalization by regulating the accessibility of the receptor to the arrestin-mediated internalization pathway.56,57 Desensitization of the β2-adrenoceptor and the LH receptor proceeds through a palmitoylated, hyperphosphorylated state. The β2-adrenoceptor contains a cAMP-dependent protein kinase consensus sequence in the close vicinity of the palmitoylation site. It is possible that reduced palmitoylation of the β2-adrenoceptor exposes the cAMP-dependent protein kinase site and causes constitutive desensitization of this receptor.
Diversity of Receptor-Ligand Binding
Different GPCRs have evolved varying molecular mechanisms for interacting with specific agonists. This structural diversity superimposed on a common seven transmembrane template reflects the conservation of an efficient protein structure for signal transduction across a membrane and the need to distinguish diverse activating ligands as different as a photon of light and a 40 kDa protein.
In general, GPCRs are activated by receptor-specific ligands that bind to their extracellular or transmembrane domains. Rhodopsin is unique in terms of activation of the receptor by the ligand. Its ligand, 11-cis-retinal, is coupled via a protonated Schiff’s base to the aldehyde moiety of retinal and the ε-amine of Lys7.43.58 This 11-cis-retinylidene moiety acts as an inverse agonist and prevents spontaneous activation of the receptor. The protonated Schiff’s base forms a salt bridge, with Glu3.28 located at the boundary between TM3 and the first extracellular loop, thus bringing TM3 and TM7 into apposition. In rhodopsin, the chromophore is buried completely inside the protein, with no accessibility to the aqueous or membrane environment. Photon absorption switches retinal from the 11-cis to the all-trans-retinylidene conformation, which neutralizes the salt bridge between the protonated Schiff’s base and Glu3.28 and activates the receptor.59
The simplest mechanism by which a ligand activates a receptor is to bind the transmembrane core.51 The protonated amine present in all biogenic amines (i.e., adrenaline, noradrenaline, dopamine, histamine, serotonin, and acetylcholine) makes direct contact with Asp3.32, a residue conserved in all aminergic receptors.60 This Asp is essential for neurotransmitter binding, but not for signal generation. In certain aminergic receptors, this interaction with the protonated amine is shared with the residue 3.36.61 In catecholamine receptors, the catechol ring of the ligand has been found to dock in the pocket between TM5 and TM6. In some neurotransmitter receptors, the meta- and para-hydroxy groups of the catecholamine agonist hydrogen bind Ser5.42 and Ser5.46, respectively.60 The cluster of aromatic residues of TM6 is highly conserved among aminergic GPCRs, and includes Trp6.48, Phe/Trp6.51, and Phe6.52. These residues have been implicated in ligand binding in many aminergic receptors.44,60
Peptide hormone receptors for short peptide ligands such as formyl receptor or gonadotropin-releasing hormone receptor (three and ten residues, respectively) complex with the peptide ligand through interactions involving both extracellular loops and the transmembrane core.62 Peptide hormone receptors with larger peptide agonists such as glucagon, parathyroid hormone, or calcitonin (30 to 40 amino acids) use both the aminoterminus and the extracellular loops to generate high-affinity ligand binding.
Receptors for thrombin and other proteases are activated by proteolysis of the aminoterminus.63 The protease ligand thrombin specifically recognizes an amino acid sequence in the aminoterminus of the receptor and cleaves it. The new shorter aminoterminus revealed by proteolysis functions as a tethered ligand that binds intramolecularly and activates the receptor.
The glycoprotein hormones, which include luteinizing hormone, follicle-stimulating hormone, chorionic gonadotropin, and thyroid-stimulating hormone, are the largest (30 to 40 kDa) and most complex GPCR agonists. These ligands are heterodimers that contain a common α subunit and a hormone-specific β subunit. The initial high-affinity binding site of these receptors is located in their large (300 to 400 residues) aminoterminus domains.
The metabotropic glutamate subclass of GPCRs is characterized by a large extracellular domain similar to bacterial periplasmic-binding proteins that contain the agonist binding site.33 The three-dimensional crystal structure of this domain has been solved for the metabotropic glutamate receptor type 1.6 This so-called Venus flytrap module consists of two lobes separated by a large cleft in which agonists bind. Another feature of subclass III receptors is that they all form dimers, either homodimers64 or heterodimers.65
The GABAB receptors are constitutive heterodimers (see later) that represent a novel principle of receptor processing and signal transduction in that two nonfunctional seven α-helical transmembrane span proteins associate to form a functional G protein–coupled receptor.66
Mechanism of Receptor Activation
The most widely accepted pharmacologic model to explain GPCR activation is the ternary complex model.67 This model has been extended to explain the observation that, under certain conditions, several GPCRs can activate G proteins in the absence of agonists.68 The extended ternary complex model proposes that the receptor exists in an equilibrium between two conformational states: the inactive (R) and the active (R*) state. In the absence of agonists, the basal level of activity of the receptor is determined by the equilibrium between R and R*. The efficacy of ligands is thought to be dependent on their ability to shift the equilibrium between these two states.69 Whereas most properties of GPCRs can be explained by the extended ternary complex model, other models have been proposed.70
When receptors are expressed heterologously in cell lines, many show spontaneous activity in the absence of agonist. Consistent with predictions of the extended ternary complex model,68 inverse agonist ligands have been identified that are able to decrease the basal level of activity of the receptors.71,72 Many ligands that previously were considered antagonists have been found to suppress this basal level of signaling and now are considered inverse agonists. According to the model, full and partial agonists bind R* with higher affinity than R, shifting the equilibrium to the activated state, whereas inverse agonists bind R with higher affinity, shifting the equilibrium to the inactive state.
The inactive state is stabilized by several intramolecular interactions, such as the salt bridge stabilizing TM3 and TM7 in rhodopsin. Similar stabilizing interactions have been suggested in the angiotensin AT1 receptor and the α1B-adrenoceptor.73 Various point mutations in many GPCRs have been found to increase the basal agonist–independent activity of the receptors.73,74 As described later, some of these activating mutations contribute to several endocrine diseases. The constraining intramolecular interactions are proposed to be released upon activation (or by specific mutations), causing key sequences to be exposed to the G protein. This hypothesis is supported by the observation that a mutation that causes constitutive activation on the β2-adrenoceptor is associated with marked structural instability and enhanced conformational flexibility.75
The crystal structure of rhodopsin has been solved at high resolution in its inactivated state, and the unliganded opsin in its G protein–interacting conformation.42,94 These results and data from cross-linking and site-directed spin labeling,76,77 together with x-ray diffraction data, suggest that activation by light opens a cleft at the cytoplasmic end of the helix bundle.59 TM7, which contains the protonated Schiff’s base linkage with the chromophore, and TM3 and 6 are critical to the activation of GPCRs.75 Photoisomerization of the chromophore neutralizes the salt bridge between the protonated Schiff’s base and its counterion Glu3.28 in TM3 (see earlier). This salt bridge corresponds to a similar ionic interaction formed between norepinephrine and an acidic side chain (3.32) in adrenergic receptors. TM7 is kinked at a highly conserved proline residue. This region of TM7 is stabilized by many interhelical constraints,31,42 including the salt bridge between the protonated Schiff’s base and Glu3.28, and an interaction with a kinked region in TM6 containing the conserved Pro6.50. Therefore, once the salt bridge is lost, a set of hydrogen bonds among TM7, TM1, and TM2 no longer would remain. These changes rearrange the TMs, especially TM2, TM6, and TM7, leading to receptor activation.
Isomerization from 11-cis to the all-trans-retinylidene induces a large displacement of the C13 methyl group. This group interacts with Trp6.48 in the ground state and, after photoisomerization, the indole ring is able to rotate during the activation process. Trp6.48 is highly conserved among GPCRs, and the binding of a ligand could induce the movement of TM6 via its indole ring.
The conformational changes described previously lead to rearrangement of the cytoplasmic side of the GPCR, allowing receptor G protein coupling. One of the proposed key events in the activation process among subclass I GPCRs involves the protonation of the Asp3.49 in the highly conserved Asp-Arg-Tyr motif at the cytoplasmic side of TM3. This “protonation hypothesis” has been supported by experiments showing that charge-neutralizing mutations, which mimic the unprotonated state of the aspartic acid, cause constitutive activation of receptor subtypes such as α1B-adrenoceptor and the β2-adrenoceptors.24 Experimental data have been supported by molecular modeling and computational simulations. Thus, the so-called arginine cage model proposed that Arg3.50 forms an ionic interaction with Arg3.49 in the inactive state of the receptor. During receptor activation, Asp3.49 becomes protonated, and the Arg3.50 side chain is released. The conserved bulky side chain of Ile3.54 restricts positioning of the arginine side chain, promoting a rearrangement that characterizes the active state of the receptor.
Interaction is observed between the 3.49 and 3.50 residues in the crystal structure of rhodopsin.42 In addition, the guanidinium group of Arg3.50 interacts with the polar side chains of the cytoplasmic side of TM6. These arrangements support the previous conclusions that protonation of the acidic group of Asp3.49 and movement of the cytoplasmic side of TM6 are critical for receptor activation.
The recent crystal structure of opsin provides additional insights into the active state of rhodopsin.51,94 The crystal structure of low pH opsin revealed major movements of TM6. The largest differences between opsin and dark rhodopsin occur at the cytoplasmic end of TM3, TM4, and TM6. It is not known how this structural change relates to the ability of the activated receptor to interact with its G protein. Although the inactive β2-adrenoceptor is structurally more similar to rhodopsin, the ionic lock is not formed between Arg3.50 and Glu6.60. Moreover, the position of TM4 is more similar to TM5 of opsin. Thus, the structure of the β2-adrenoceptor appears to be in an intermediate conformation, which may explain its basal activity toward Gs. The adenosine A2A-ligand–bound structure48 suggests that there is no general, family-conserved receptor-binding pocket in which selectivity is achieved through different amino acid side chains. Rather, the pocket itself can vary in position and orientation and so may offer increased opportunity for receptor diversity and ligand selectivity. Refining the model for the pathway from agonist binding to G protein activation will require a combination of crystal structures, including GPCR–G protein complexes.
Receptor/G Protein Coupling and Selectivity
Although many GPCRs also have been found to couple via G protein–independent mechanisms,11 the interaction with heterotrimeric G proteins is the major signaling mechanism of these proteins. G protein activation modulates classical downstream effectors, such as adenylate cyclases,78 phospholipases,79,80 and ionic channels,81 through well-characterized molecular mechanisms.82,83
HETEROTRIMERIC G PROTEINS
G proteins were described for the first time in the 1970s by Rodbell84,85 and Gilman.86 The “nucleotide binding protein necessary to reconstitute the stimulation of adenylate cyclase” was first purified by Gilman’s laboratory.87
Two main families of signal transduction–related proteins that bind and hydrolyze guanine nucleotides comprise monomeric G proteins (small G proteins) and heterotrimeric G proteins. Monomeric G proteins are approximately 200 amino acids in size. They are implicated in biological processes such as cell cycle, protein secretion, or intracellular vesicular interaction, and they present certain structural similarities with heterotrimeric G proteins.88 Heterotrimeric G proteins have three subunits (α, β, and γ).10,89 The β and γ subunits form a dimer that does not dissociate under physiologic conditions. Heterotrimeric G proteins are soluble intracellular proteins that may associate with the plasma membrane through covalent attachment of fatty acids. The Gα subunit may be myristoylated or palmitoylated, and the Gγ subunit may be farnesylated or geranylgeranylated.
G proteins are classified according to the Gα subunit present in the heterotrimeric complex. Mammals have more than 20 different α subunit subtypes encoded by 17 genes. Some subtypes are different isoforms. The Gα subunits form four different families (Gαs, Gαi, Gαq, and Gα12) based on the degree of homology of the primary structure (Table 5-2). These subtype sequences are highly conserved across different species. In this regard, no differences are evident in the amino acid sequences between mouse and human Gαs.
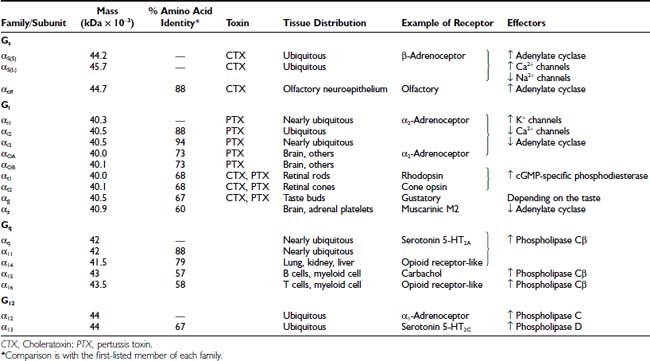
Except for the G proteins that are expressed in sensory organs (Gαt, Gαg, and Gαolf) and certain subtypes found mainly in hematopoietic (Gα16) or nervous (Gαo) cells, most α subunits are ubiquitous. Moreover, each cellular type generally expresses four or five different α subunits. The G proteins can be expressed at high concentration in certain tissues. Thus, in brain, Gαo may represent 1% to 2% of the total membrane protein.
Six different subtypes of Gβ subunits (35 to 39 kDa) have been reported in mammals and are encoded by six different genes. Twelve different Gγ subunits, which are encoded on 12 different genes, show a high heterogeneity. Although in theory these protein subtypes could form 72 different Gβγ dimers, not all combinations are expressed in vivo. The potential functional importance of such Gβγ diversity is not yet understood.90 Most Gβγ dimers (except for β1γ1 expressed in the retina) appear to exhibit similar functional properties.
The high-resolution structure of the heterotrimeric G proteins Gt and Gi shows the overall shape of the guanosine diphosphate (GDP)-bound heterotrimer and residues on the surface that can interact with other proteins. The α subunit contains three domains. The guanosine triphosphatase (GTPase) domain is highly conserved and has structural features in common with monomeric G proteins. The nucleotide binding pocket, as well as sites for binding the receptor effector and Gβγ, are located within the GTPase domain. The helical domain surrounds the nucleotide binding pocket and has been proposed to increase the affinity of guanosine triphosphate (GTP) binding, to act as a tethered GTPase activating protein (GAP, see later), and to participate in effector recognition. The third domain is the aminoterminus of the Gα subunit, which forms an α helix.
The tertiary structure of the β subunit consists of two domains. The aminoterminal domain forms an α helix of about 20 amino acids. The carboxyterminal domain Gβ contains the so-called β-propeller fold, formed by seven antiparallel β-sheets. This motif is formed by a class of repeating sequences (WD) also found in a variety of other proteins; many of them are unrelated to members of the Gβ family.
The γ subunit contains two α helices connected by a four amino acid loop. In the Gβγ dimer, the aminoterminals of Gβ and Gγ show helix-helix interactions, and the carboxyterminal of Gγ is embedded on one surface of the toroidal Gβ subunit. This structure explains the stability of the Gβγ dimer.
The G proteins undergo a cyclic activation and deactivation process, which transmits the signal from receptor to effector (Fig. 5-3).10 When an agonist activates the GPCR, the GDP-bound Gαβγ heterotrimer interacts with the receptor, and the α subunits decrease the affinity for GDP. Because the concentration of GTP is higher than that of GDP in the cytoplasm, GDP is displaced by GTP in the nucleotide binding pocket. Once GTP is bound, the now active α subunit dissociates from both the receptor and Gβγ. This active state of the Gα persists until its intrinsic Mg2+-dependent GTPase activity hydrolyzes GTP to GDP. All isoforms of Gα are GTPases, but the intrinsic rate of GTP hydrolysis varies from one type to another. Once GTP is cleaved to GDP, the Gα and Gβγ reassociate and become inactive until stimulated by a receptor (see Fig. 5-3).
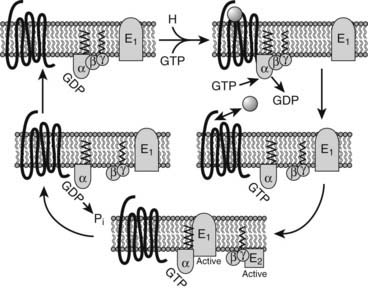
FIGURE 5-3. G protein cycle. In the inactive state, the heterotrimeric guanosine diphosphate (GDP)-bound G protein is associated with the receptor (top right). Activation of the receptor leads to guanosine triphosphate (GTP)/GDP exchange in the Gα subunit, dissociation of the Gα and Gβγ subunits from the receptor and each other, and interaction with signaling effector proteins (E1,E2). The intrinsic guanosine triphosphatase (GTPase) activity of Gα regenerates the GDP-bound Gα subunit, which reassociates with Gβγ and the receptor, thereby completing the cycle.
The original hypothesis about the G protein–mediated signal transduction was that GTP-bound Gα subunits were able to activate effectors, while Gβγ dimers were only negative modulators. Thus, release of free Gβγ from a highly expressed G protein such as Gi can deactivate other stimulatory Gα subunits such as Gαs. This view changed with the discovery that Gβγ, as well as Gα, could positively regulate effectors such as K+ channels.91
MOLECULAR BASIS OF RECEPTOR/G PROTEIN COUPLING
Most GPCRs can recognize and activate only a limited set of the many structurally similar G proteins (as defined by their α subunits) expressed in a cell.10,89 Based on this G protein coupling preference, GPCRs can be subdivided broadly into Gi/o-, Gs-, and Gq/11-coupled receptors (see Table 5-1). Although it has been reported that several GPCR subtypes can activate G12/13 proteins, receptors that preferentially activate G12/13 proteins have not yet been identified.
As discussed earlier, receptor activation is likely to involve a cleft opening at the cytoplasmic receptor surface, which enables the receptor to expose previously buried residues critical for G protein recognition and activation. All GPCR intracellular loops have been implicated in receptor/G protein specificity.
The length of the intracellular loop 1 is highly conserved among GPCRs, suggesting an important structural role in G protein coupling. Several studies have shown that the structural integrity of the intracellular loop 1 is critical for receptor/G protein coupling. However, it remains to be determined whether these residues are in direct contact with the G protein, or whether indirect conformational effects are exerted by intracellular loop 1 on the accessibility of other regions (such as intracellular loops 2 and 3) that can interact directly with the G protein.
Mutational studies on several subclass I GPCRs have demonstrated that replacement of the arginine residue within the highly conserved Glu/Asp-Arg-Tyr motif at the aminoterminus of the intracellular loop 2 (see earlier) abolishes or drastically reduces G protein coupling.92 However, charge-conserving mutations result only in modest reduction of receptor function. These results suggest that the conserved arginine residue interacts with an electron-rich site on the G protein. In this regard, both N-formyl peptide receptor and rhodopsin have been shown to be unable to associate physically with G proteins when the conserved arginine residue was mutated. Thus, this arginine residue has been proposed to represent the primary trigger for the release of GDP from the receptor/G protein complex.
The intracellular loop 2 has been implicated in the regulation of receptor/G protein coupling selectivity through the use of GPCR chimeras.93 Several studies have reported that substitution of the intracellular loop 2 (alone or together with other intracellular receptor regions such as the intracellular loop 3 or the carboxyterminus) from a donor receptor into a functionally different receptor can confer on the receptor chimera the G protein coupling profile of the donor receptor. In this context, the intracellular loop 3 also has been involved in receptor/G protein coupling selectivity. However, it usually is not sufficient for controlling all the coupling characteristics of a particular receptor. Thus, detailed mutational analysis of different muscarinic receptors (M1 through M5) has identified single amino acids, primarily hydrophobic or noncharged, within the intracellular loop 3 that play a key role in determining receptor/G protein coupling selectivity in conjunction with residues present in the intracellular loop 2.
As discussed earlier, the helical structure of the carboxyterminus that forms a fourth intracellular loop is one of the most striking findings of the high-resolution structure of GPCRs.51 Recent findings with the crystal structure of the active conformation of opsin and an 11 amino acid synthetic peptide derived from the extreme C terminus of the transducing Gαt subunit provide insight into the structural changes involved in signal transfer from the receptor to the G protein.94 It seems that G protein and ligand-binding sites are coupled in the active receptor conformation, fitting in with the classical receptor theory, in which the active conformation is stabilized by the ligand and/or the G protein.
REGULATION OF RECEPTOR/G PROTEIN COUPLING BY RNA EDITING
It has been demonstrated that the messenger RNA (mRNA) for the serotonin (5-HT)2C receptor is posttranscriptionally modified.95 The mRNA sequences that encode adenosine are converted to inosines by the action of double-stranded RNA (dsRNA) adenosine deaminases. This editing generates multiple receptor isoforms, some of which have been found to differ in their signaling properties. It has been demonstrated that in rat brain, at least seven 5-HT2C receptor isoforms show distinct anatomical distributions. In human brain, 14 different 5-HT2C receptor isoforms involving editing of the second intracellular loop have been reported.
The generation of different 5-HT2C receptor isoforms in several brain regions may control specific cellular responses. In this regard, both receptor/G protein coupling96,97 and receptor activation of effectors98,99 have been reported to differ for edited 5-HT2C receptor isoforms. Moreover, the conformation of the 5-HT2C receptor isoforms studied by computer structural approaches suggests that the edited second intracellular loops would differ structurally.100 RNA editing also has been found to regulate 5-HT2C receptor transactivation of the small G protein RhoA.101 5-HT2C receptor isoforms also differ in their desensitization and trafficking.102
EFFECTS OF POSTTRANSLATIONAL MODIFICATIONS ON RECEPTOR/G PROTEIN COUPLING SELECTIVITY
The selectivity of receptor/G protein coupling has been shown to be regulated by receptor phosphorylation.103 Activation of the β2-adrenoceptor stimulates an increase in intracellular cAMP via activation of Gs proteins, as well as activation of protein kinase A (PKA) and stimulation of mitogen-activated protein kinases (MAPKs) through a pathway involving Gi proteins. Studies with mutant β2-adrenoceptors that lack PKA phosphorylation sites have shown that receptor-mediated activation of Gi proteins is dependent on phosphorylation of the β2-adrenoceptor by PKA. As receptor phosphorylation by PKA decreases the coupling efficiency of the β2-adrenoceptor to Gs proteins, phosphorylation represents a switch mechanism for regulating G protein–coupling selectivity.
As we have discussed earlier, many GPCRs include a conserved cysteine residue in their carboxyterminus that may be modified covalently by palmitic acid. Some receptors, such as endothelin receptor, have been shown to regulate G protein coupling selectivity through this palmitoylation.
REGULATORS OF G PROTEIN SIGNALING PROTEINS (RGS PROTEINS)
The rate of inactivation of G proteins by intrinsic GTP hydrolysis is augmented by regulator of G protein signaling (RGS) proteins.104–106 RGS proteins are a family of highly diverse proteins that share a conserved 120 amino acid domain (RGS domain). The RGS domain binds directly to the activated Gα-GTP subunits and acts as a GTPase activating protein (GAP). Thus, RGS proteins accelerate GTP hydrolysis, attenuating or modulating hormone and neurotransmitter receptor-mediated responses (Fig. 5-4).
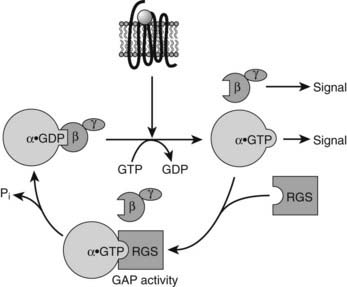
FIGURE 5-4. Schematic illustrating the role of regulator of G protein signaling (RGS) proteins in accelerating guanosine triphosphatase (GTPase) activity and inactivation of active Gα subunits. GAP refers to GTPase-activating protein.
More than 20 different RGSs have been described in mammals that can be grouped into five subfamilies (RZ, R4, R7, R12, and RA) based on sequence similarities (Table 5-3). Several G protein effectors and regulators such as G protein–coupled receptor kinases (GRKs), phospholipase C–beta (PLC-β), RhoGEF, and cyclic guanosine monophosphate (cGMP) phosphodiesterase also display GAP activity and have domains distantly related to the RGS domain (see Table 5-3). The globular and mostly helical structure of the RGS domain has been solved by x-ray crystallography in the RGS4-Gαi1 complex.

RGS proteins also have been implicated in the modulation of adenylate cyclase, MAPK, IP3/Ca2+ signaling, K+ conductance, and visual signaling. Larger RGS domain–containing proteins have additional domains that are likely to contribute to cellular functions, in addition to attenuating G protein activation.104 For example, the guanine-nucleotide-exchange factor for the monomeric small G protein RhoA (RhoGEF) induces the GTP for GDP interchange in the small G protein and contains an RGS domain. When RhoGEF binds the receptor-activated Gα13, the GEF activity of RhoGEF increases, leading to RhoA activation and modulation of several downstream effectors.
Most of the RGS proteins are predicted to be cytosolic and are recruited to the plasma membrane by activated Gα subunits. RGS proteins are widely expressed, and many tissues express multiple RGS proteins. Mechanisms identified that contribute to the selectivity of RGS proteins for specific GPCR signaling pathways are their cell type expression and intracellular localization, the timing of their expression, and the presence of other domains outside of the RGS domain that interact with other signaling proteins. The central, specific, and diverse signaling effects of RGS proteins make them important potential targets for drug development.104,107
ACTIVATORS OF G PROTEIN SIGNALING (AGS)
The activation of heterotrimeric G proteins in the absence of involvement of a typical heptahelical GPCR has been described.108 Three activators of G protein signaling (AGS) proteins have been identified with the use of a functional screen based on the pheromone response pathway in Saccharomyces cerevisiae. AGS proteins could provide novel mechanisms for input to G protein signaling pathways.
G Protein–Dependent Effectors
Signaling via the large family of GPCRs can lead to many cellular responses, ranging from regulation of intracellular levels of cAMP to stimulation of gene transcription. Members of the GPCR family have been grouped into different categories dependent on the particular G protein subtypes with which they predominantly interact. Thus, receptors that couple to Gs proteins will stimulate adenylate cyclase in many cells, and Gq/11-coupled receptors can mobilize intracellular Ca2+ via activation of phospholipase C. Signaling by GPCRs is ascribed conventionally to activation of heterotrimeric G proteins. However, over the past several years, an increasing number of examples have been described in which such receptors signal in situations in which G protein activation appears to be minimal or absent.11,13
ADENYLYL CYLASE SIGNALING
Up to now, at least nine closely related isoforms of adenylyl cyclase (AC1 through AC9) and two splice variants of AC8 have been cloned and characterized in mammals.78 They present large sequence homology in the primary structure of their catalytic sites and the same predicted three-dimensional structure. Each AC isoform and variant consists of two hydrophobic domains (with six transmembrane spans) and two cytoplasmic domains, resulting in a pseudosymmetrical protein. The cytoplasmic domains (C1 and C2), which constitute the catalytic site, are subject to intracellular regulations specific for each subtype. In addition to their ability to respond to Gαs, the different isoforms can receive signals from a variety of sources, including other G proteins (e.g., Gαi, Gβγ), protein kinases (PKA, PKC, and calmodulin), phosphatases (calcineurin), and calcium, and these isoforms are able to support and integrate differential regulatory pathways through cross-talk with other signal transduction systems.
PHOSPHOLIPASE C SIGNALING
Receptor-dependent activation of PLC and consequential activation of inositol lipid-signaling pathways constitute some of the physiologic effects of many extracellular stimuli. Multiple PLC isozymes were purified and subsequently cloned.79 These initially included PLC-β, PLC-γ, and PLC-δ (an earlier PLC-α proved not to be a functional phospholipase) classes of isozymes. The mammalian family has expanded in the last decade to include PLC-ε, PLC-ζ, and PLC-η. These proteins exhibit relatively low overall homology, but each PLC isozyme contains conserved catalytic core regions historically designated as the X and Y domains. The intervening sequence between X and Y domains is approximately 50 to 100 amino acids in most isozymes but contains two Src homology 2 (SH2) domains and a single Src 3 (SH3) domain in PLC-γ isozymes. With the exception of PLC-γ, all PLC isozymes contain a plekstrin homology (PH) domain, which potentially binds membrane phosphoinositides or regulatory proteins.
The four isoforms of PLC-β (β1 through β4) are stimulated directly and independently by Gα-GTP and Gβγ to hydrolyze PI(4,5)P2 and trigger inositol lipid signaling.80 Multiple studies have demonstrated that Gαq family members differ in their efficacy for PLC-β activation, depending on the PLC-β isoform. It is interesting to note that Gα16 is a more effective activator of PLC-β2 than are Gαq, Gα11, and Gα14. Like Gα15/16, PLC-β2 is expressed specifically in hematopoietic cells, and preferential activation of this PLC isotype by Gα15/16 among other Gαq family members suggests that these proteins are functionally linked in native cells. Except for the retina-specific PLC-β4, the remaining PLC-β subtypes both are widely distributed, as are Gαq and Gα11. Collectively, these findings indicate that Gαq family members may couple selectively to PLC-β isoforms in native cells to generate tissue- or receptor-specific responses in vivo.
ION-CHANNEL SIGNALING
Ion channels, encoded by several hundreds of genes in humans, differ widely in molecular structure, selectivity to ions, and mechanisms of operation.81 In spite of their diversity, these proteins share a general structural motif: a pore formed by and enclosed within the transmembrane segments of the channel protein, through which ions transverse the plasma membrane. Most mammalian ion channels are regulated by neurotransmitters and hormones via GPCRs. Both Gα and Gβγ subunits can regulate ion channels indirectly (via second messengers and protein kinases) or directly, via physical interaction between G protein subunits and the channel protein. Although direct modulation by G proteins has been proposed for many ion channels, it has been firmly established for only two families: (i) some voltage-activated Ca2+ channels, which are inhibited by Gβγ, and (ii) G protein–activated inwardly rectifying K+ channels, which are activated by Gβγ. Additional proteins play distinct roles in positioning Gβγ or regulating its effect or gating. The main role of Gα is to provide the specificity of signaling and to serve as Gβγ donors, but its role could extend beyond these functions. Several other ion channels appear to be modulated by direct interactions with G proteins, but additional studies are needed before this is firmly established.
G Protein–Coupled Receptor Signaling Networks
In the classical model of GPCR signaling, receptor activation induces dissociation of heterotrimeric G proteins into α- and βγ-subunits. These subunits sequentially activate effector molecules, including second messenger systems, such as adenylate cyclases78 or phospholipases80 and ion channels.81 These linear pathways lead to the modulation of various well-characterized cellular responses (see Table 5-2; Fig. 5-5).82,83 However, the number of identified effectors is considerably smaller than the large number of GPCRs. Because most of the cells express multiple types of GPCRs that signal through a relatively limited number of effectors, it is not surprising that signal processing involves cross-regulation among the different signaling pathways. Moreover, the discovery of new GPCR-activated heterotrimeric G protein–independent signaling pathways demonstrates that the classical paradigm of linear G protein signaling pathways represents only part of a more complex receptor-regulated signaling system.
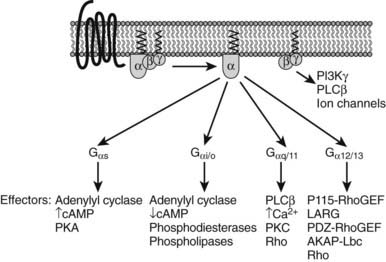
FIGURE 5-5. Diversity of heterotrimeric G protein signaling. Heterotrimeric G proteins are divided into four subfamilies (see Table 5-2). Each G protein activates several downstream effectors.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


