FIGURE 67-1. Osteopetrosis. This teenage girl with Albers-Schönberg disease has a “rugger-jersey” spine typical of adult (“benign”) osteopetrosis. The vertebral end plates are markedly thickened.
Skeletal scintigraphy reveals fractures and osteomyelitis.24 Magnetic resonance imaging (MRI) helps assess patients undergoing bone marrow transplantation, because engraftment restores medullary spaces.25
Laboratory Findings
In infantile OPT, serum calcium concentrations depend largely upon gastrointestinal calcium absorption, because of the impaired bone resorption.26 Secondary hyperparathyroidism with elevated serum levels of calcitriol is common.26 Hypocalcemia can occur, especially with achlorhydria, and causes rickets.23
In adult OPT, biochemical indices of mineral homeostasis usually are unremarkable, although serum PTH levels can be increased.27
Serum acid phosphatase and the brain isoenzyme of creatine kinase often are elevated and seem to originate from the dysfunctional osteoclasts.28
Histopathologic Findings
Failure of osteoclast action provides a pathognomonic finding in OPT6—primary spongiosa synthesized during endochondral bone formation persists as “islands” of calcified cartilage encased within trabecular bone. Osteoclast numbers are increased, normal, or, rarely, decreased. In infantile OPT, these cells are usually abundant.29 Their nuclei are especially numerous, but the “ruffled borders” and “clear zones” that characterize functioning osteoclasts are absent.30 Fibrosis often crowds marrow spaces. Adult OPT shows increased amounts of osteoid, and osteoclasts can be few and may lack ruffled borders, or they can be especially numerous and large.31
Cause and Pathogenesis
Most patients with OPT have diminished osteoclast-mediated acidification at sites of bone resorption due to defects in CA II, the α3 subunit of the vacuolar proton pump, or chloride channel 7 (CLCN7).32 Heterozygous loss-of-function mutation within CLCN7 causes Albers-Schönberg disease.33 Homozygous or compound heterozygous CLCN7 mutations lead to severe or intermediate OPT.33 Malignant OPT usually is due to deactivating mutations in the gene TCIRG1 (ATP6i), which encodes the α3 subunit of the vacuolar proton pump.34 Defects in the “gray-lethal” and OSTM1 genes cause especially severe OPT.35 OL-EDA-ID represents disruption of an essential modulator of NF-κB.21 Recently, loss-of-function mutations within the genes that encode the receptor activator of nuclear factor-κB (RANK) or its ligand (RANKL) were discovered in especially rare forms of autosomal recessive OPT.36,37
Ultimately, impaired skeletal resorption in OPT causes both myelophthisis and bone fragility resulting from the presence of fewer collagen fibrils interconnecting osteons.11
Treatment
Because the cause and pathogenesis, pattern of inheritance, and prognosis for the various forms of OPT can differ, a precise diagnosis is crucial before therapy is attempted. For example, infants or young children with CA II deficiency can have radiographic features of malignant OPT, yet sequential studies may show gradual resolution of bony sclerosis.6 Until recently, the patient’s family and investigation into the severity and progression of the disorder were the principal considerations. Now, diagnosis has been advanced greatly by mutation analysis.38
Bone Marrow Transplantation
Bone marrow transplantation from HLA-identical donors has improved remarkably some patients with infantile OPT.32 However, this procedure is not always appropriate6 (e.g., RANKL deficiency)36 because the pathogenetic defect must be corrected by entry of donor cells into the osteoclast lineage.32
Because severely crowded medullary spaces appear less likely to engraft, early intervention is best.39 Use of marrow from HLA-nonidentical donors warrants continued study. Purified progenitor cells in blood from HLA-haploidentical parents have been useful.39a Marked hypercalcemia can occur as osteoclast function begins.40
Dietary and Medical Therapy
Some success has been reported when a calcium-deficient diet is given. Conversely, calcium supplementation may be necessary for symptomatic hypocalcemia or rickets.23 Large oral doses of calcitriol together with dietary calcium restriction (to prevent absorptive hypercalciuria/hypercalcemia) sometimes improves infantile OPT.41 Calcitriol may stimulate defective osteoclasts, but resistance can occur.41 Long-term infusion of PTH helped one infant,42 perhaps by enhancing calcitriol synthesis. Diminished leukocyte production of superoxide serves as the basis for recombinant human interferon-γ-1b treatment for severely affected children.41
High-dose glucocorticoid treatment stabilizes pancytopenia and hepatomegaly. One case report describes inexplicable reversal of malignant OPT after prednisone therapy alone.43 Prednisone and a low-calcium/high-phosphate diet may be effective.44
In CA II deficiency, the RTA has been treated with bicarbonate supplementation, but the long-term impact is unknown. Bone marrow transplantation corrects the OPT and slows cerebral calcification of CA II deficiency, but does not alter the RTA.45
Supportive Therapy
Hyperbaric oxygenation can be important for osteomyelitis of the jaw.6 Surgical decompression of the optic and facial nerves may benefit some patients. Joint replacement can be helpful.46 Internal fixation may be necessary for femoral fractures.47
Early prenatal diagnosis of OPT by ultrasound generally has been unsuccessful. Conventional radiographs occasionally reveal malignant OPT late in pregnancy. However, mutation analysis is now available in commercial laboratories and can be used to detect OPT in utero.38
PYCNODYSOSTOSIS
Pycnodysostosis was discovered in 1962.48 Most reports have come from the United States or Europe, but its prevalence seems greatest in Japan.49 Parental consanguinity with autosomal recessive inheritance explains ≈30% of cases. In 1996, loss-of-function mutation of the gene that encodes cathepsin K was identified.50
Clinical Features
Pycnodysostosis is diagnosed during infancy or early childhood because of disproportionate short stature and a relatively large cranium with fronto-occipital prominence, small facies and chin, beaked nose, high-arched palate, obtuse mandibular angle, dental malocclusion with retained deciduous teeth, proptosis, and bluish sclera.51 The anterior fontanel and cranial sutures remain open. Mental retardation affects ≈10% of cases.51 Hands are small and square and fingers are short and clubbed from acro-osteolysis or aplasia of terminal phalanges. Pectus excavatum may occur. Recurrent fractures typically involve the lower limbs and cause genu valgum deformity, although patients usually walk independently. Adult height ranges from 4 ft 3 in to 4 ft 11 in. Recurrent respiratory infections and right heart failure from upper airway obstruction caused by micrognathia trouble some patients.
Radiographic Findings
Pycnodysostosis shares many features with OPT.22 Both cause generalized osteosclerosis and recurrent fractures. The osteosclerosis first appears in childhood, is uniform, and increases with age. The calvarium and the skull base are sclerotic, and orbital ridges are dense. Although long bones have narrow medullary canals, the striking modeling defects of OPT do not occur. Endobones and radiodense striations are absent.22 Other distinguishing findings in pycnodysostosis include delayed closure of cranial sutures and fontanels (prominently the anterior), obtuse mandibular angle, wormian bones, gracile clavicles that are hypoplastic laterally, and hypoplasia or aplasia of the distal phalanges and ribs.52 Hypoplasia of facial bones, sinuses, and terminal phalanges is characteristic. Vertebral bodies are dense with anterior and posterior concavities, but transverse processes are uninvolved. Lumbosacral spondylolisthesis is not uncommon, and lack of segmentation of the atlas and axis may be noted.22
Laboratory Findings
Anemia is not a concern. Serum calcium and inorganic phosphate levels and alkaline phosphatase activity usually are normal. Cortical bone appears unremarkable except for diminished osteoblastic and osteoclastic activity.53 Abnormal inclusions have been described in chondrocytes. Electron microscopy suggests that degradation of collagen is defective.53
Cause and Pathogenesis
Deactivating mutations in the gene that encodes cathepsin K cause pycnodysostosis.50 Cathepsin K is a lysosomal cysteine protease that is highly expressed in osteoclasts.54 Hence, impaired collagen degradation is a fundamental defect. The rate of bone accretion and the size of the exchangeable calcium pool seem reduced. Bone remodeling and therefore quality are compromised.55 Accordingly, pycnodysostosis can be thought of as a form of OPT.
Additionally, killing activity and interleukin-1 secretion by circulating monocytes is compromised.56 Virus-like inclusions have been reported in osteoclasts.57 Defective growth hormone secretion and low serum insulin-like growth factor 1 levels have been described.58
Treatment
No medical therapy is recognized. Bone marrow transplantation has not been reported.
The orthopedic challenges have been reviewed briefly.59 Long bone fractures typically are transverse and heal at a satisfactory rate, but delayed union and massive formation of callus can occur. Intramedullary fixation of long bones is formidable because of their hardness.
Extraction of teeth is difficult, and mandibular fracture has occurred.51 Osteomyelitis of the mandible may require antibiotic and surgical treatment.
PROGRESSIVE DIAPHYSEAL DYSPLASIA (CAMURATI-ENGELMANN DISEASE)
Progressive diaphyseal dysplasia (PDD) is an autosomal dominant disorder that affects all races. The condition was described by Cockayne in 1920.60 Camurati discovered its heritable nature.61 Engelmann characterized the severe, typical form in 1929.61 In 2001, mutations that are activating defects were identified in the gene that encodes transforming growth factor (TGF)-β1.62
Characteristically in PDD, painful hyperostosis occurs gradually on both periosteal and endosteal surfaces of long bones.22 However, the clinical and radiographic expression is quite variable.63 In severe cases, osteosclerosis is widespread, including the skull and axial skeleton. Some carriers have no radiographic changes, but bone scintigraphy is abnormal.
Clinical Presentation
PDD typically presents during childhood with limping or a broad-based and waddling gait, leg pain, muscle wasting, and diminished subcutaneous fat in the extremities mimicking muscular dystrophy.64 However, severely affected patients also have a characteristic body habitus that includes an enlarged head with prominent forehead, proptosis, and thin limbs with thickened bones. Cranial nerve palsies can develop when the skull is affected. Puberty sometimes is delayed. Raised intracranial pressure may occur. Palpable bony thickening, skeletal tenderness, and sometimes hepatosplenomegaly are present, as well as Raynaud’s phenomenon and other findings suggestive of vasculitis.65 Radiologic studies typically show progressive disease, but the course is variable and symptom remission sometimes occurs during adult life.
Radiologic Features
Hyperostosis of major long bone diaphyses, the principal finding, represents proliferation of new bone on both periosteal and endosteal surfaces.22 Shafts of long bones gradually widen and develop irregular surfaces. Sclerosis is fairly symmetrical and spreads to involve metaphyses, but the epiphyses are characteristically spared (Fig. 67-2). Tibias and femurs most often are involved, less frequently the radii, ulnae, humeri, and, occasionally, the short tubular bones. Clavicles, scapulae, and the pelvis also may become thickened. Age of onset, rate of progression, and degree of bony involvement are highly variable. With mild disease, scintigraphic abnormalities may be confined to the lower limbs. Maturation of the new bone increases the hyperostosis. However, in severely affected children, some skeletal areas may appear osteopenic.
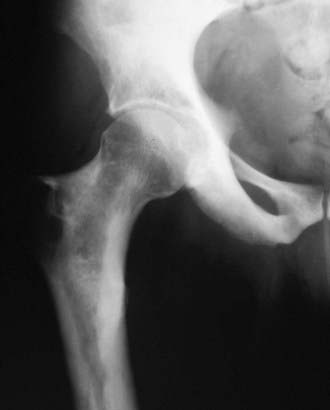
FIGURE 67-2. Progressive diaphyseal dysplasia. This man with Camurati-Engelmann disease has irregular hyperostosis (cortical thickening) of the proximal femur that characteristically does not extend to the end of the long bone.
Clinical, radiographic, and scintigraphic findings are generally concordant.66 Occasionally, however, bone scans are unremarkable despite marked radiographic changes. This seems to reflect advanced but quiescent disease. Increased radioisotope accumulation with few radiographic alterations can represent early skeletal involvement.
Laboratory Findings
Routine biochemical parameters of bone and mineral homeostasis typically are normal, although serum alkaline phosphatase levels can be elevated. Mild hypocalcemia and significant hypocalciuria may occur in severe disease, likely reflecting positive calcium balance.67 Mild anemia and leukopenia and an elevated erythrocyte sedimentation rate seem to reflect the poorly characterized systemic disturbances.65
Histopathology shows new bone formation along diaphyses. Disorganized woven bone undergoes maturation and then incorporation into the cortex. Electron microscopy of muscle has revealed myopathic changes and vascular abnormalities.64
Cause and Pathogenesis
The clinical and laboratory features of severe PDD and its responsiveness to glucocorticoid treatment indicate an inflammatory connective tissue disease.65 Now, the disorder is known to involve mutations in a specific region of the gene that encodes TGF-β1. Consequently, a “latency-associated peptide” encoded by this gene remains bound to TGF-β1, keeping this enhancer of bone formation active.62 Aberrant differentiation of precursor cells to osteoblasts has been discussed as a pathogenetic mechanism.68
Treatment
PDD is a chronic and somewhat unpredictable disorder. Symptoms may remit during adolescence or adult life. Glucocorticoid therapy (typically small doses of prednisone on alternate days) can relieve bone pain and improve histologic abnormalities in bone.69 Bisphosphonate therapy may increase skeletal pain.70
ENDOSTEAL HYPEROSTOSIS
In 1955, van Buchem et al.71 described hyperostosis corticalis generalisata. Subsequently, additional forms of endosteal hyperostosis were characterized.72 The hallmark of these disorders is thickening of cortical bone primarily on endosteal surfaces.22
Van Buchem disease is an autosomal recessive condition71 that is considerably rarer than the number of case reports might suggest.73 The principal clinical feature is progressive asymmetrical enlargement of the jaw during puberty. The mandible becomes markedly thickened with a wide angle, but no prognathism is noted. Dental malocclusion is uncommon. Affected individuals may be symptom free, but cranial sclerosis also occurs and recurrent facial nerve palsy, deafness, and optic atrophy from narrowing of cranial foramina are common and can develop during infancy. Long bones may hurt with applied pressure but are not fragile.71 Endosteal cortical thickening leads to homogenously dense diaphyses with narrowed medullary canals. However, long bones are shaped properly. Osteosclerosis also affects the skull base, facial bones, vertebrae, pelvis, and ribs.22 Serum alkaline phosphatase from bone may be increased, but calcium and inorganic phosphate levels are unremarkable.
Sclerosteosis, like van Buchem disease, is an autosomal recessive disorder that occurs primarily in people of Dutch ancestry.72 However, sclerosteosis differs from van Buchem disease in that patients are excessively tall and have syndactyly.72 At birth, only fused fingers may be noted.73a Syndactyly reflects cutaneous or bony fusion of the middle and index fingers. During early childhood, skeletal overgrowth involves especially the skull and causes facial disfigurement. Progressive bone thickening widens the jaw, resulting in prognathism.74 Patients become tall and heavy. Deafness and facial palsy are prominent problems. A small skull vault may increase intracranial pressure, causing headaches and compressing the brain stem.75 Intelligence is normal. Life expectancy can be shortened.76 Long bones become widened as cortices thicken. Vertebral pedicles, ribs, and the pelvis may become dense. Fusion of ossicles and narrowing of the internal auditory canals and cochlear aqueducts may occur.72 Enhanced osteoblast activity with failure of osteoclasts to compensate explains the dense bone of sclerosteosis.75 No abnormality of calcium homeostasis or of pituitary function has been documented.77 No specific medical treatment is available. Surgical correction of syndactyly is difficult if there is bony fusion. Management of the neurologic dysfunction has been reviewed.75
Deactivating mutations in the gene that encodes sclerostin (SOST) cause sclerosteosis,78,79 whereas van Buchem disease results from a 52-kb deletion that diminishes a downstream enhancer of SOST.80 Sclerostin binds to LRP5/6, antagonizes canonical Wnt signaling,81 and promotes the apoptosis of osteoblasts.82 Accordingly, sclerostin deficiency in sclerosteosis and van Buchem disease enhances bone formation.
An autosomal dominant (Worth) type83 of endosteal hyperostosis is relatively benign and was rediscovered recently as the “high bone mass phenotype.”84 Some affected individuals have torus palatineus.85 Certain domain-specific, gain-of-function mutations of LRP5 encoding low-density lipoprotein receptor–related protein 5 cause this form of increased skeletal mass.86 LRP5 activation may decrease the biosynthesis of systemic serotonin, leading to enhanced bone formation.87 Despite excesses of good quality bone, the condition is not always benign, as cranial nerve palsies, skeletal pain, and oropharyngeal exostoses may occur.88
OSTEOPOIKILOSIS
Osteopoikilosis (spotted bones) is usually a radiographic curiosity transmitted as an autosomal dominant trait with a high degree of penetrance.89 However, joint contractions and limb length inequality can occur, especially with accompanying radiographic changes of melorheostosis. When connective tissue nevi called dermatofibrosis lenticularis disseminata are present, this is the Buschke-Ollendorff syndrome.90 The osteopoikilosis is asymptomatic, but if misunderstood can lead to studies for metastatic disease, etc.91 Hence, family members at risk should be counseled. Radiographs show numerous small foci of usually round or oval osteosclerosis22 involving the ends of the short tubular bones, the metaepiphyseal regions of the long bones, and the tarsal, carpal, and pelvic bones (Fig. 67-3). These are thickened trabeculae or islands of cortical bone and do not change appearance over decades. Radionuclide accumulation is not increased on bone scanning.91
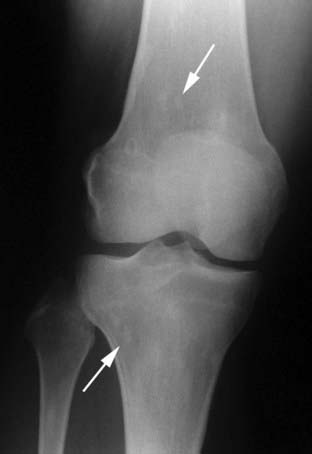
FIGURE 67-3. Osteopoikilosis. Faint, round or oval areas of osteosclerosis (arrows) are present in the metaphases of the distal femur and proximal tibia.
The nevi usually appear before puberty, involve the lower trunk or extremities, and are small asymptomatic papules or yellow or white disks or plaques, deep nodules, or streaks.90 They represent excessive, unusually broad, and markedly branched elastin fibers in the dermis.90
Heterozygous deactivating mutations in the LEMD3 gene cause osteopoikilosis and the Buschke-Ollendorff syndrome.92 LEMD3 is an inner nuclear membrane protein that antagonizes TGF-β1 and bone morphogenetic protein signaling.
OSTEOPATHIA STRIATA
Osteopathia striata features linear striations at the ends of long bones and in the ileum22 and is a curiosity if the skeletal findings occur alone as an autosomal dominant trait. However, osteopathia striata also occurs in clinically important syndromes. These include osteopathia striata with cranial sclerosis93 due to WTX mutations,94 where cranial nerve palsies are common.93 Osteopathia striata with focal dermal hypoplasia (Goltz syndrome) is a serious X-linked dominant disorder featuring widespread linear areas of dermal hypoplasia through which adipose tissue herniates, together with bony defects in the limbs.95
Gracile striations affect trabecular bone, especially the metaepiphyses of major long bones and the periphery of the iliac bones.22 Lesions are unchanged for years. Radionuclide accumulation is not increased during bone scanning.91
PACHYDERMOPERIOSTOSIS
Pachydermoperiostosis (hypertrophic osteoarthropathy: primary or idiopathic) causes clubbing of the digits, hyperhidrosis, and thickening of the skin, especially involving the face and forehead (cutis verticis gyrata). Clubbing, periostitis, and pachydermia constitute the classic triad of features. However, some patients have just one or two of these findings. Radiographs reveal periosteal new bone formation, especially in the distal limbs. Autosomal dominant inheritance with variable expression is established,96 but autosomal recessive transmission also seems to occur.97
Blacks seem to be affected more often than whites, and men more severely than women. Presentation is typically during adolescence, but variable.96,97 Symptoms emerge over a decade, but then can abate.98 Progressive enlargement of the hands and feet may cause a “paw-like” appearance. Palms may be wet. Arthralgias of the elbows, wrists, knees, and ankles are common. Acro-osteolysis has been reported. Stiffness of the appendicular and axial skeleton can develop. Compression of cranial or spinal nerves has been described. Cutaneous changes include coarsening, thickening, furrowing, pitting, and oiliness of especially the scalp and face, with some affected individuals described as “acromegalic.” Fatigue is common. Myelophthisic anemia with extramedullary hematopoiesis may occur.
Radiologic Features
Severe periostitis thickens the distal portions of tubular bones—especially the radius, ulna, tibia, and fibula. Clubbing is obvious, and acro-osteolysis can occur. Ankylosis of joints, especially in the hands and in the feet, may trouble older patients.22 The principal diagnostic challenge is secondary hypertrophic osteoarthropathy (pulmonary or otherwise), but this presents a smooth, undulating appearance.99 In pachydermoperiostosis, periosteal proliferation is more exuberant and irregular and often involves epiphyses. Bone scanning in either condition reveals symmetrical, diffuse, regular uptake along the cortical margins of long bones, especially in the legs, causing a “double-stripe” sign.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


