FIGURE 26-1. Height velocity chart for boys aged 0 to 19 years. Centiles 3 to 97 illustrated with 50th centile in bold. Shaded zone, Variation in timing of pubertal growth spurt. Visually, the chart depicts the rapid but rapidly decelerating growth during the first 4 years of life followed by a much slower declination until the onset of the pubertal growth spurt.
(Copyright Castlemead Publications.)
Growth hormone is the main mediator of postnatal growth,2 and virtually any chronic childhood illness will modify secretion. As such, care needs to be exercised in the evaluation of GH secretion in these situations. Although GH deficiency (GHD) may be considered a form of IGF deficiency,3 this approach may be limited. Since GH receptor and post-receptor issues and GHD in adults are considered elsewhere, this chapter will focus primarily on GHD as related to disorders of the hypothalamo-pituitary axis in children.
History
The history of GHD starts with the pursuit of therapeutic interventions which antedate attempts to measure serum concentrations of GH. In 1932, a treatment to promote growth with a crude anterior pituitary extract was reported,4 but it was not until Raben’s observations of 19585 and the general availability of methods of GH extraction that large studies could be conducted. These larger studies showed a beneficial effect of human GH in promoting growth in children with clear physical signs suggestive of GH deficiency.6–9
Growth hormone immunoassays postdated the initial therapeutic studies of GH.10 The advent of radioimmunoassay allowed the measurement of the concentration of GH in blood in response to a variety of pharmacologic stimuli, thereby paving the way to a better understanding of which children might benefit from the then limited supplies of GH. This limitation in supply largely dictated clinical research until the advent of unlimited supplies of biosynthetic human GH (r-hGH) resulting from bioengineering technology in the late 1970s and early 1980s.
Our understanding of the physiology of GH secretion stemmed from the pioneering work of Geoffrey Harris and his group in Oxford, who suggested that release of GH from the pituitary was under the control of a releasing factor secreted from the hypothalamus. The demonstration by Brazeau et al.11 of a GH release-inhibiting factor (somatostatin) led to a radical change in the thinking around the control of GH secretion. The final demonstration of a GH stimulating factor came in 1982 when GH releasing hormone was isolated and characterized from two pancreatic tumors.12,13 During the search for the releasing factor, little was made of a further stimulating factor described by Bowers et al., which although synthetic in nature, formed the basis from which GH releasing substances and their receptors were identified. Finally the natural ligand, ghrelin, was isolated from the stomach.14
For a considerable period of time, GH was believed to act via the generation of a further endocrine factor from the liver, somatomedin-C or IGF-I.15 Further work led to the realization that liver was not the only source of IGF-I, and in a series of classic experiments, Green16 and Isaksson17 demonstrated in adipose tissue and cartilage, respectively, that IGF-I was generated locally and acted in a paracrine manner to promote clonal expansion of the cell population.
Epidemiology
The reported incidence of GHD is to a large extent dependent on the criteria employed to establish the diagnosis and reflects the wide variation in the stringency of diagnostic testing. In one U.K. study, an incidence of 1 in 60,000 live births was reported,18 although a survey of Scottish schoolchildren led to a calculated prevalence of 1 per 4000 live births,19 a value similar to that of the Utah Growth Study (1 in 3480 live births).20
Several large surveys have indicated that approximately 25% of children diagnosed with GHD have an underlying “organic” cause for their condition, such as trauma, CNS tumors, inflammation, irradiation, or anatomic abnormalities of the hypothalamus or pituitary.21,22 The remainder are labeled as “idiopathic” GHD. Such surveys are likely to overestimate the number of true cases of idiopathic GHD because of variation in the diagnosis of GHD. Recent advances in developmental endocrinology suggest that many patients labeled previously as idiopathic GHD have genetic abnormalities or subtle anatomic abnormalities affecting the hypothalamus, pituitary, or both.
Pathogenesis
A list of causes of GHD is provided in Table 26-1. As mentioned already, “idiopathic” GHD constitutes by far the largest group of patients, although advances in developmental biology are forcing a rethink in this area.
Table 26-1. Causes of GH Deficiency
| Congenital |
| Genetic: See Table 26-2 |
| Associated With Structural Defects of the Brain: |
| Associated With Midline Facial Defects: |
| Idiopathic |
| Acquired |
| Trauma: |
| Infection: |
| CNS tumors: |
| Following Cranial Irradiation |
| Following Chemotherapy |
| Pituitary Infarction |
| Neurosecretory Dysfunction |
| Transient: |
GENETIC AND STRUCTURAL ABNORMALITIES
Pituitary Development
The pituitary gland, which consists of anterior, intermediate, and posterior lobes, is a central regulator of growth, metabolism, and development. Its complex functions are mediated via hormone-signaling pathways that act to regulate the finely balanced homeostatic control in vertebrates by coordinating signals from the hypothalamus to peripheral endocrine organs (thyroid, adrenals, and gonads). The mature anterior pituitary gland is populated by five neuroendocrine cell types defined by the hormone produced: corticotropes (corticotropin [formerly adrenocorticotropic hormone, ACTH]), thyrotropes (thyroid-stimulating hormone [TSH]), gonadotropes (luteinizing hormone [LH], follicle-stimulating hormone [FSH]), somatotropes (GH) and lactotropes (prolactin [PrL]).23 The posterior gland secretes vasopressin and oxytocin. The origins of the anterior and posterior lobes of the pituitary gland are embryologically distinct. Rathke’s pouch, the primordium of the anterior pituitary, arises from the oral ectoderm, whereas the posterior pituitary derives from neural ectoderm. Development of the anterior gland follows a similar pattern in a number of different species but has been best studied in rodents.
In the mouse, anterior pituitary development occurs in four distinct stages: pituitary placode formation; the development of a rudimentary Rathke’s pouch; the formation of a definitive pouch; and finally the terminal differentiation of the various cell types in a temporally and spatially regulated manner (Fig. 26-2). The apposition of Rathke’s pouch and the diencephalon, which later develops into the hypothalamus, is maintained throughout the early stages of pituitary organogenesis24 and appears to be critical for normal anterior pituitary development. A number of signaling molecules—fibroblast growth factor-8 (Fgf8),24–26 bone morphogenetic protein 4 (Bmp4),24,25 and Nkx2.126—that are expressed in the neural ectoderm and not in Rathke’s pouch are thought to play a significant role in normal anterior pituitary development, as illustrated by the phenotype of mouse mutants that are either null or hypomorphic for these alleles. These signaling molecules activate or repress key regulatory genes encoding transcription factors such as Hesx1, LIM homeobox 3 (Lhx3), and LIM homeobox 4 (Lhx4) within the developing Rathke’s pouch that are essential for subsequent development of the pituitary.23,24
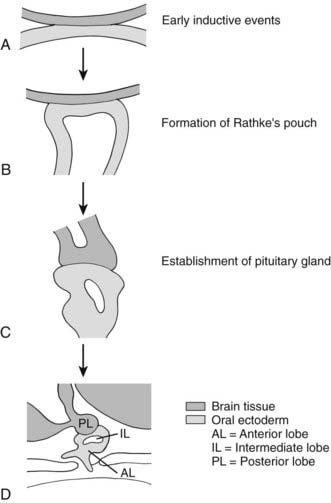
FIGURE 26-2. Formation of the pituitary gland. Four-stage process commencing with early inductive events as the infundibulum of the diencephalon abuts the roof of the oral cavity. Pituitary established with signaling gradients generating spatial defined patterns of gene expression and specific cell lineages in the definitive gland.
(Reproduced from Valette-Kasic and Enjalbert, Topical Endocrinology, February 2003.)
The final stage of pituitary gland development entails the terminal differentiation of the progenitor cells into the distinct cell types found within the mature pituitary gland. This process is tightly regulated by extrinsic factors (Fgf8, Bmp2, Bmp4, and Bmp7) that emanate from the surrounding infundibulum and the juxtapituitary mesenchyme. These then establish gradients of transcription factors (Lhx3, Six3, prophet of Pit1 [Prop1], Pit1, Nkx3.1, Islet-1 [Isl1], Lhx4, Six1, Brain-4 [Brn4], and pituitary forkhead [Pfrk]).25,26 These genetic gradients lead to a wave of cell differentiation. Each of the five anterior pituitary cell types differentiates in a temporally and spatially regulated manner (Fig. 26-3),29–32 and this process is dependent upon a number of transcription factors such as Pit1, T-pit, and steroidogenic factor 1 (Sf1).33,34
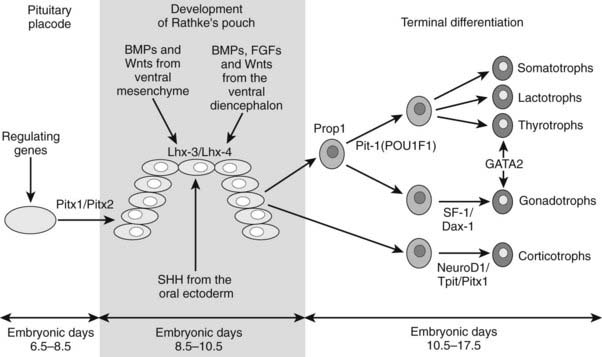
FIGURE 26-3. Temporal sequence of events of pituitary development in the mouse.
(Reproduced from Valette-Kasic and Enjalbert, Topical Endocrinology, February 2003.)
Less is known about pituitary development in humans, but it appears to mirror that in the rodent. Spontaneous or artificially induced mutations in the mouse have led to significant insights into human pituitary disease, and identification of mutations associated with human pituitary disease have in turn been invaluable in defining the genetic cascade responsible for the development of this complex structure.
Disorders of Pituitary and Extra-Pituitary Development in Humans
A number of genetic abnormalities have been identified in children who were previously thought to have idiopathic GHD or combined pituitary hormone deficiency (CPHD)35–37 (Table 26-2). In some cases, extra-pituitary manifestations may be associated. Mutations within the paired-like homeobox gene HESX1 are associated with the phenotypes of GHD, CPHD, and septo-optic dysplasia, a condition characterized by forebrain, pituitary, and eye abnormalities such as optic nerve hypoplasia.38,39 The inheritance and phenotypes are variable, with both dominant and recessive modes of inheritance described. Intriguingly, HESX1 mutations are classically associated with anterior pituitary hypoplasia with an undescended posterior pituitary and an absent or thin infundibulum.40
Table 26-2. Genes Implicated in Isolated Growth Hormone Deficiency and Combined Pituitary Hormone Deficiencies
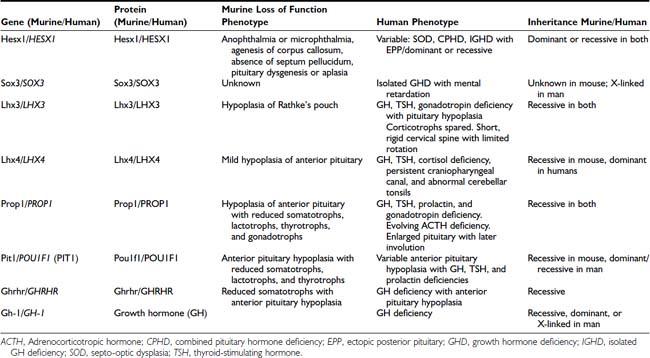
Mutations within the LIM-domain genes LHX3 and LHX4 are associated with CPHD with extrapituitary manifestations such as a short neck and steep cervical spine in the case of LHX341 and an abnormal cerebellum in the case of LHX4.42 The inheritance of LHX3 mutations is recessive, whereas that associated with LHX4 mutations is dominant, unlike the murine phenotype.
Mutations within the gene encoding the transcription factor Sox2 is associated with hypopituitarism in the mouse and humans. SOX2 is one of the earliest known genes to be expressed in embryonic stem cells and neural progenitors. Although mutations in the mouse are associated with a generalized reduction in all pituitary cell types, in the human, the most frequent pituitary defect is hypogonadotropic hypogonadism, with GHD less frequent. Other features include severe eye defects, esophageal atresia, hypothalamic hamartomata, learning difficulties, and sensorineural hearing loss.43,44
Recent studies with SOX3 suggest a possible explanation for the predominance of males in many series of GHD. SOX3 is sited on the X chromosome (Xq26-27) and appears to be important not only in pituitary development but is also associated with mental retardation.45 These observations of GHD and brain developmental abnormalities are particularly important because neurodevelopmental handicap has often been ascribed to untreated neonatal hypoglycemia, whereas structural developmental problems may be a more pertinent explanation.
It is clear that our understanding of the etiology of hypopituitarism is rudimentary. The mechanisms whereby mutations in the genes that have been identified to date lead to a particular phenotype are largely unknown. Additionally, many cases of hypopituitarism may be due to changes in regulatory regions of known genes or perhaps within novel genes that have yet to be identified.
Growth Hormone–Releasing Hormone and Its Receptor
Because growth hormone–releasing hormone (GHRH) and its receptor (GHRHR; GHD Type 1B) are critical to somatotroph population expansion, abnormalities in either are likely to be associated with severe GHD. No mutations of the human GHRH gene have been identified, but mutations in GHRHR have been identified in a number of pedigrees.46 Two large pedigrees have been identified in Pakistan (Glu72Stop mutation)47 and in northeastern Brazil (donor splice mutation in position 1 of intron 1; IVS1, G-A, +1).48 All patients reported to date have been either homozygous or compound heterozygous for mutations of the GHRHR gene. Serum GH concentrations fail to rise following standard provocative testing, as well as after GHRH administration. The patients resemble the little mouse (lit/lit), which has a mutation of the GHRH receptor gene affecting the ligand-binding domain (Asp60Gly; D60G).49
Somatotroph Development
A number of mouse models exist in which somatotrope development has been impaired. These include the Ames, the Jackson, and the Snell dwarf mice. A missense point mutation within the prophet of Pit1, or PROP1 gene (S83P), has been shown to be responsible for the Ames dwarf mouse.50 The phenotype results from a failure of initial determination of the Pit1 lineage required for production of GH, PrL, and TSH. The Ames pituitary gland contains less than 1% of the normal complement of somatotrophs and decreased numbers of lactotrophs and thyrotrophs. In humans, mutations within the transcription factor Prop1 are associated with CPHD in the form of GH, prolactin, TSH, and gonadotropin deficiency.51,52 A proportion of individuals with PROP1 mutations will develop cortisol deficiency.53 Additionally, a number of individuals with mutations within PROP1 develop transient pituitary masses with subsequent involution (Fig. 26-4).54 The exact mechanism underlying this phenomenon remains unclear, but it is clearly important to exclude mutations within PROP1 in patients with pituitary “tumors” especially the nonfunctional variety. There is considerable variability in the timing of the endocrinopathy, and a number of patients will actually commence puberty but then arrest halfway through. Recently, a mutation within PROP1 was identified in a patient who actually achieved a normal final height without receiving any GH treatment. PROP1 mutations are thought to be the commonest cause of familial CPHD and are usually recessive.
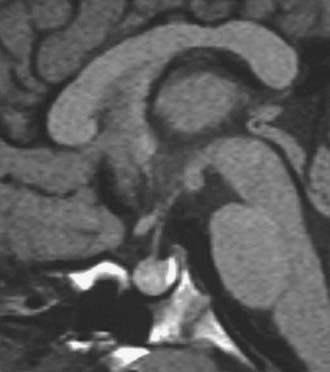
FIGURE 26-4. Pituitary “tumor” in patient with PROP-1 mutation. Saggital MRI scan of the pituitary revealing a large, globular anterior pituitary with normal posterior pituitary enhancement. Subsequent scans revealed involution of the mass and resultant empty sella.
Pit1 (now known as Pou1f1)55,56 is a member of the POU family of homeodomain proteins and contains a highly conserved bipartite DNA-binding domain consisting of the POU homeodomain, required for low-affinity DNA binding, and a POU-specific domain, responsible for the specificity of DNA binding and potential interactions with other proteins. The Snell dwarf mouse, characterized by pituitary hypoplasia and GH, PrL, and TSH deficiencies, has a point mutation (W261C) within the Pit1 gene, affecting the third helix of the POU homeodomain. This abrogates binding of Pit1 to its target promoter sequences. Several mutations and deletions of the POU1F1 gene have been identified in humans with CPHD, characterized by the combination of GH, PrL, and TSH deficiency.57,58 Mutations have been described which separately affect the DNA-binding capacity of POU1F1 or its transactivation properties. Autosomal dominant transmission, resulting from a dominant negative effect, has been observed in mutations affecting dimerization of POU1F1, transactivation (P24L), or in the relatively common R271W mutation, which results in increased binding to promoter elements and disruption of transcriptional activation.59 Autosomal recessive transmission is found with other mutations, such as A172stop, E250stop, R143G, A158P, and P239S.60 Variability in phenotype has been reported, although most patients exhibit growth retardation during the first year of life. GH and PrL deficiency is complete; TSH secretion may be observed during infancy but declines progressively during the early months of life. Magnetic resonance imaging (MRI) scanning revealed a marked variability in the size of the anterior pituitary, with some patients demonstrating a normal pituitary and others having a hypoplastic pituitary. After appropriate GH and thyroxine replacement, patients appear to enter puberty normally and have normal fertility. Lactation may be impaired. In some patients, TSH secretion may be normal.61
GH1 Gene
The GH1 gene, located at chromosome 17q22-24, is part of a cluster of five structurally related genes: GH1, CSHP (chorionic somatomammotropin pseudogene), CSH (chorionic somatomammotropin), GH2 (or placental variant), and CSH2. Mutations within the GH1 gene are associated with isolated GH deficiency (Table 26-3). Large, recessive, inherited deletions are associated with absence of GH protein (Type 1A GHD). Complete loss of pituitary GH secretion occurs secondary to deletions resulting from nonhomologous crossing over at different sites in the GH and chorionic somatomammotropin (CS) gene cluster. The most common deletion is 6.7 kb, but deletions of 7.0, 7.6, and greater than 45 kb have also been observed.62 Wagner et al.63 have also described the GHD IA phenotype in a patient with a point mutation in the GH signal peptide, E23X, resulting in premature termination of translation. Patients typically have an excellent initial response to GH therapy, but because of the absence of a normal GH molecule in fetal life, an attenuation of the growth response to exogenous GH may result from the development of anti-GH antibodies,64 although this event has been described less frequently with newer GH preparations.
Table 26-3. Genetic Abnormalities in GH1
| A. | Type 1 | Type 1A. Autosomal recessive GHD due to total absence of GH synthesis. |
| Type 1B. Autosomal recessive GHD due to splicing defects in GH1 or defects in GHRHR genes. | ||
| B. | Type 2 | Autosomal dominant GHD due to splice-site and missense mutations in the GH1 gene, resulting in dominant negative expression of the GH1 gene. Abnormal folding of mutant interferes with storage and secretion. |
| C. | Type 3 | X-linked GHD. Xq21-q22 with X-linked agammaglobulinemia and Xq22-q27 with X-linked mental retardation. |
Type IB GHD is due to homozygous splice-site mutations within the GH1 gene or homozygous mutations within the GHRHR. It is associated with an excellent response to GH treatment, with no formation of antibodies.
Type II GHD is autosomal dominant and associated with splice-site mutations.65 These mutations lead to the production of two alternatively spliced GH molecules, 20 and 17.5 kD hGH. Mutations in an exon splice enhancer within exon 3 of the GH1 gene have also been associated with autosomal dominant GHD.66 The generation of the 17.5 kD form of hGH has a dominant negative effect and prevents the secretion of the normal wild-type 22kD hGH, with a consequent deleterious effect on pituitary somatotropes. In a murine model of this dominant negative mutation, there is loss of somatotroph number67 and progressive damage to adjacent pituitary cells (with later failure of PrL, TSH, and gonadotropin secretion).68
Seven different splice-site mutations have been reported to date. In addition, three missense mutations (R183H, P89L, and V110F) were also recently implicated in IGHD Type II. These patients have a normal GH1 allele but are unable to secrete the normal form of GH in appropriate concentrations. The mutant protein therefore exerts a dominant negative effect. As in the mouse evolution of other hormonal deficiencies, including ACTH, TSH, and gonadotropin, deficiencies have been described in patients with some dominant GH1 mutations.69
GHD Type III, an X-linked form of isolated GH deficiency (IGHD), has been reported in patients with hypogammaglobulinemia. To date, no alteration in the GH1 gene has been identified in this condition, and the genetic mechanisms remain unknown. Recently a polyalanine expansion within the transcription factor Sox3, which lies at Xq26-27, has been associated with X-linked GHD and mental retardation.45 Intriguingly, duplications of this region of the X chromosome have been associated with X-linked panhypopituitarism, and SOX3 has been implicated as the gene associated with this phenotype.70,71
Bioinactive Growth Hormone Molecule
Since the GH molecule exists in multiple molecular forms resulting from alternative splicing or posttranslational processing, some cases of short stature have been hypothesized to be the consequence of abnormal ratios of the various GH forms.72 The first report of two individuals heterozygous for point mutations in the GH gene was described by Takahashi et al. and detailed the biochemistry and molecular genetics.73 The mutant GH molecules (R77C and D112G) were capable of binding to the GH receptor, perhaps even with increased affinity, but were unable to stimulate tyrosine phosphorylation of GH-activated intracellular signaling intermediates in a normal manner. The ability of the R77C mutant to behave in a dominant negative manner was demonstrated by its ability to inhibit the in-vitro actions of wild-type GH.
Structural Abnormalities
In addition to the structural abnormalities associated with the genetic problems described above, GHD can occur in the setting of other cranial or midline abnormalities such as holoprosencephaly, nasal encephalocele, single central incisor, and cleft lip and palate.
As methods of radiologic evaluation of the CNS have improved, an increasing percentage of patients with idiopathic GHD have been identified to have structural abnormalities.74–80 Many of these are associated with some of the genetic abnormalities described above, but the findings are worthy of separate consideration. In particular, the finding on MRI of an undescended (erroneously called “ectopic”) posterior pituitary (PPE) was commoner in males than females (3:1 when PPE present versus 1:1 if normal anatomy) in patients with CPHD as compared with IGHD (49% versus 12%), breech delivery (32% versus 7%), and associated congenital brain anomalies (12% versus 7%).
These findings appear to be best explained by a defect in induction of the mediobasal structure of the brain in the early embryo rather than the product of birth trauma, as previously suggested.77 Whether pituitary insufficiency is the result of hypothalamic or pituitary dysgenesis or the product of hypoplasia or sectioning of the pituitary stalk is not always clear. Perinatal problems, however, including breech presentation, may prove to be the consequence rather than the cause of underlying CNS abnormality. The concept that PPE, stalk section or hypoplasia, and pituitary hypoplasia may represent abnormal embryonic development rather than the consequences of birth trauma is supported by the finding of similar anatomic abnormalities in patients with septo-optic dysplasia, type I Arnold-Chiari syndrome, holoprosencephaly, and increasingly in patients with mutations in the genes controlling pituitary development.
In the empty sella syndrome, abnormalities of the sellar diaphragm allow herniation of the suprasellar subarachnoid space into the region of the sella turcica.81 This may result in damage to the sella, including the pituitary. Empty sella syndrome may be the consequence of surgery or irradiation, or may be idiopathic. It is often found in patients with mutations in PROP-1, when it may have been preceded by a pituitary mass.
ACQUIRED DEFECTS
Destructive Lesions of the Hypothalamus and Pituitary
A wide range of destructive lesions involving the hypothalamus or pituitary may present with isolated GHD or CPHD. Birth trauma, associated with abrupt delivery, prolonged labor, or extensive use of forceps, has been associated frequently with subsequent hypothalamic or pituitary dysfunction.82,83 An increased incidence of GHD has been reported in breech deliveries, although it is still unclear whether such deliveries lead to acquisition of pituitary dysfunction or, on the other hand, whether preexisting CNS abnormalities result in higher rates of abnormal birth presentations.
Tumors
Central nervous system tumors are an important cause of isolated GHD and CPHD and must be excluded in every child with GHD who does not have an obvious alternative explanation for growth failure. Midline brain tumors include germinomas, meningiomas, gliomas, colloid cysts of the third ventricle, ependymomas, and optic nerve gliomas. GHD or CPHD may also occur from local extension of tumors affecting the head or neck, such as craniopharyngeal carcinomas and lymphomas.
The major pediatric tumor involving the pituitary is craniopharyngioma, which is probably an evolving congenital malformation which develops from remnants of Rathke’s pouch.84 It accounts for 5% to 15% of intracranial tumors in childhood and 80% of tumors in the hypothalamo-pituitary region. Arising from rests of squamous cells at the embryonic junction of the adenohypophysis and neurohypophysis, it forms an enlarging cyst filled with degenerating cells, leading to cyst fluid or calcification but never to malignant degeneration (Fig. 26-5). These calcifications may be seen at times on skull films and constitute an important diagnostic sign. Although craniopharyngiomas represent the consequences of a congenital malformation, they may present clinically at any age. Significant growth failure may be observed at the time of diagnosis, but patients most commonly present with complaints of increased intracranial pressure, such as headaches, vomiting, and oculomotor disturbances; visual field defects are frequently noted at the time of diagnosis.84 Deficiency of at least one pituitary hormone, most commonly GH or gonadotropin, is present in 50% to 80% of patients. Diabetes insipidus is reported in 25% to 50% of patients at diagnosis.84,85
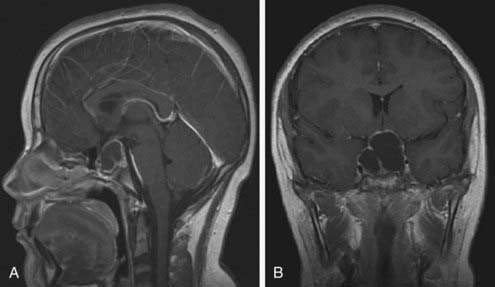
FIGURE 26-5. MRI of cystic craniopharyngioma. Saggital MRI scan (A) revealing large multicystic craniopharyngioma arising from the pituitary fossa and extending up to hypothalamus. Coronal section (B) of same lesion delineating upward and lateral spread. Both images are T-1 weighted, gadolinium-enhanced scans.
Langerhans’ cell histiocytosis may also present at any age. Langerhans cell histiocytosis (LCH) is characterized by clonal proliferation and accumulation of abnormal dendritic cells that can affect either a single site or many systems, causing multi-organ dysfunction. In children, the median age of diagnosis ranges between 1.8 and 3.4 years.86 LCH infiltrates the hypothalamo-pituitary area in 15% to 35% of patients, with subsequent development of at least one pituitary hormone deficiency.87 In a multicenter French national study of 589 pediatric patients with LCH, 145 patients (25%) had pituitary dysfunction. In 60 patients, pituitary involvement was already present at the time of diagnosis, and in 20 of them, it was the first manifestation of the disease. Patients at high risk of pituitary involvement seem to be those with multi-system disease involving skull and facial bones, mastoid, sinuses, and mucous membranes (i.e., gums, ear, nose, and throat region). Furthermore, compared to patients without pituitary involvement, patients with pituitary involvement have a higher rate of relapse (10% at 5 years versus 4.8% at 5 years) and a higher incidence of neurodegenerative LCH.88
Diabetes insipidus is the most frequently reported permanent consequence of LCH and the commonest endocrinopathy; almost all patients with pituitary involvement have DI. The second commonest endocrinopathy is GHD, which occurs in 14% of all patients with LCH and in more than 40% of patients who have pituitary involvement.87 In the vast majority of patients, GHD is associated with DI, with a median interval of 2.9 to 3.5 years between the diagnosis of DI and development of GHD. Isolated GHD, or the association of GHD with other anterior pituitary hormone deficiencies, occurs less commonly.
Pituitary MRI findings in patients with LCH include thickening of the pituitary stalk, suggestive of the infiltrative process enhancing changes in the pituitary gland and hypothalamus, and absence of the bright signal of the posterior pituitary in T1-weighted images, caused by the loss of the phospholipid-rich ADH secretory granules. The latter is an invariable feature of patients who develop DI.89 Although at the time of diagnosis of DI, 75% show a thickened pituitary stalk, only 24% have persistent stalk thickening after 5 years. These changes are variable and do not correlate with treatment or with clinical recovery; DI persists in all cases.
Long-term follow up of patients with LCH has shown that the already established hormone deficiencies cannot be reversed by treatment.89 Recently, however, isolated case reports have suggested that treatment with the purine analogue 2-chlorodeoxyadenosine (2-CDA) may reverse established DI.90 Subsequent studies of this form of therapy, used in refractory cases of LCH involving the CNS, showed that 2-CDA may result in partial or complete radiologic improvement of the mass lesion, but the endocrine consequences of the disease, including DI and panhypopituitarism, do not reverse.91 Patients treated with the JLSG-96 protocol who have been followed up for 5 years developed DI with an incidence of 3.1% to 8.9%, depending on the extension of the disease (single system multisite versus multisystem).92
Radiotherapy used for the treatment of LCH is within the dose range of 10 to 15 Gy, which is known to be unlikely to cause GHI. However, radiotherapy has been associated with an increased risk of GHD despite the fact that the dose was less than 15 Gy, a finding that may reflect the severity and extent of the disease rather than the direct effect of radiotherapy.
Irradiation of the Central Nervous System
Cranial irradiation used for the therapy of solid brain tumors and as prophylaxis for leukemia can lead to abnormal hypothalamo-pituitary function. The sensitivity of the HP axis to radiation depends upon the dose, fractionation, tissue location, and the age of the patient.93 Such damage is typically difficult to assess precisely, since the hypothalamus and pituitary may differ in the extent of involvement, and the loss of function may evolve with time. Sensitivity to CNS radiation may differ among patients, although the majority of children will experience some degree of hypothalamic or pituitary dysfunction within 5 years of receiving 30 Gy.94 GHD also occurs with doses of 18-24 Gy,95 and subtle dysfunction may be observed at even lower doses. GH secretion generally appears to be the most sensitive to irradiation, followed by TSH, gonadotropins, and finally ACTH. This may relate to the unique position of the GHRH neurons on the surface of the hypothalamus and not deep within the structure as previously thought.96
Pituitary dysfunction evolves over several years following irradiation, so such children should be monitored for growth deceleration. Provocative GH testing may be within normal limits, but measures of spontaneous GH secretion frequently demonstrate abnormalities.97 Serum concentrations of IGF-1 or insulin-like growth factor–binding protein-3 (IGFBP-3) may not be reduced in the early years following cranial irradiation.98
Cranial irradiation may also result in precocious puberty, leading to an early pubertal growth spurt, advanced skeletal maturation, and ultimately reduced stature. This may be superimposed upon any growth restriction that results from the spinal irradiation for the primary problem.99 Low-dose irradiation is frequently associated with a precocious onset of puberty; higher doses may result in gonadotropin deficiency and pubertal delay. In the irradiated child with early puberty, therapy with gonadotropin-releasing hormone (GnRH) analogues should be considered, with or without GH treatment, to delay epiphyseal fusion.
Lower doses of radiation (24 Gy) are also associated with GHD in approximately 30% to 60% of cases. Craniospinal irradiation used in the treatment of posterior fossa tumors and total body irradiation used in conditioning regimens for bone marrow transplant are also associated with damage to the epiphyses, with subsequent disproportionate short stature.
Traumatic Brain Injury
Traumatic brain injury (TBI) has been recognized as a cause of acquired hypopituitarism in a number of adult studies. Data on pediatric patients are sporadic, but TBI is probably underdiagnosed.100 These effects may be significant, considering the scale of the problem. In the United Kingdom, 180 children per 100,000 population per year sustain a head injury, with 5.6 per 100,000 requiring intensive care and almost one third of those admitted to ICU undergoing neurosurgery.
Although the pituitary gland is protected within the bony cavity of the sella turcica, the rich vascular network of the hypothalamus and pituitary and the structure of the pituitary stalk make it vulnerable to the effects of traumatic brain injury. The hypothalamus and pituitary have a complex vascular supply consisting of an arterial supply via the superior and inferior hypophyseal arteries from the internal carotid artery, as well as long hypophyseal vessels and a rich network of portal capillaries that surround the pituitary and infundibulum. The pathophysiology of hypopituitarism related to TBI is not clearly defined, but it is thought that it is the result of direct trauma or of vascular injury resulting in ischemia and infarction,101 an observation supported by the anatomical findings of autopsies following head trauma which include anterior lobe necrosis, pituitary fibrosis, hemorrhage, infarction, or necrosis of the pituitary stalk.102
Hormone deficiencies may be identified in the first days to weeks posttrauma (acute phase) or may develop over time (late effect). Because there is overlap between the symptoms and signs of hypopituitarism and those of neurologic/psychologic sequelae of TBI, it is possible that late evolving or partial deficiencies can remain undiagnosed for extended periods.
In the acute phase, alterations in the endocrine function may reflect an adaptive response to acute illness. The clinically significant alterations involve mainly the regulation of fluid and electrolyte balance (diabetes insipidus, SIADH, cerebral salt wasting) and the hypothalamo-pituitary adrenal axis. Most of the pituitary hormone changes observed in the acute phase are transient, and their development cannot predict the development of permanent hypopituitarism.103
Pituitary hormone deficiencies present in the acute phase are usually transient, but they may persist or appear and evolve over time. In adults, the incidence of permanent hypopituitarism ranges between 23% to 69%, depending on the study. The GH axis is the most frequently affected (10% to 33%), followed by the gonadal (8% to 23%), adrenal (5% to 23%), and thyroid (2% to 22%) axis. The prevalence of permanent DI varies between 0% and 6%.
Until recently, there were only sporadic reports of hypopituitarism following TBI in children, but prospective studies designed to address the problem in the pediatric and adolescent population are in progress. The incidence of hypopituitarism is reported to range from 10% to 60%, and although this is lower in children as compared with adults, it is not uncommon.104 In general, the long-term outcome of TBI seems to be more favorable in children, although quality-of-life issues and minor disability may persist. The extent to which endocrine dysfunction contributes to these outcomes has yet to be defined.
Growth hormone deficiency appears to be the main endocrine manifestation, followed by gonadotropin deficiency. GHD can present as growth failure, whereas delayed or arrested puberty and secondary amenorrhea may present in adolescents and in patients in the transition phase. In a number of case reports, central precocious puberty has also been described in association with head injury, presenting 0.4 to 1.6 years after the event.105
Patients with hypopituitarism after head injury may have no clinical signs and symptoms suggestive of this disorder; its correct identification requires a high degree of suspicion. A consensus guideline on the screening of patients post TBI suggests that all patients who had TBI, regardless of its severity, should undergo baseline endocrine evaluation 3 and 12 months after the event or discharge from ITU.106
Infiltrative and Inflammatory Disorders
Infiltrative diseases are uncommon causes of GHD in the pediatric population, but pituitary insufficiency may be observed secondary to CNS involvement in tuberculosis,107 sarcoidosis,108 or toxoplasmosis. Inflammation associated with bacterial, viral, fungal, or parasitic disease may also result in hypothalamic-pituitary dysfunction. Lymphadenoid hypophysitis has also been reported.
Thalassemia is a hereditary disorder characterized by quantitative defects in synthesis of globin chains that result in ineffective erythropoiesis and, in its more severe forms, transfusion dependence. The majority of complications are the consequence of the toxic effects of iron which is deposited in organs of the reticuloendothelial system, the heart, and all target organs of the endocrine system, including the pituitary.109 The anterior pituitary is very sensitive to iron overload, resulting in defective GH secretion, reduced responsiveness of GH to GHRH, and hypogonadotropic hypogonadism. The gonadotroph cells seem to be particularly vulnerable to the toxic effects of iron deposition, which may be related to the way iron is transported in cells. Failure of pubertal development and growth impairment are the most prominent endocrine complications and may occur despite early initiation of chelation therapy. It is estimated that 56% of thalassemic patients have at least one endocrinopathy; almost half have hypogonadism (40% to 59%), and 33% to 36% manifest growth failure.110 The growth impairment is the result of a number of factors that include chronic anemia and tissue hypoxia, overchelation due to the toxic effects of desferrioxamine on spinal cartilage, GH insufficiency, and possible GH insensitivity.
Psychosocial Dwarfism
Psychosocial dwarfism is a form of poor growth associated with bizarre eating and drinking behavior, social withdrawal, delayed speech, and on occasion other evidence of developmental delay.111 Periodic hyperphagia is associated with decreased GH responsiveness to standard provocative stimuli but also with subnormal responses to exogenous GH therapy. Removal from the stressful environment, which usually involves removal from the home, is accompanied by a restoration of normal GH secretion, typically within weeks, and a period of catch-up growth.112,113 The mechanisms for this reversible form of GHD are unclear, but it is of note that a variety of psychiatric conditions in adults may be associated with decreased spontaneous and provocative GH secretion. Establishing the diagnosis of psychosocial dwarfism requires documentation of catch-up growth and restoration of normal GH secretion following correction of the environmental situation.
Clinical Features
THE HYPOTHALAMO-PITUITARY-SOMATOTROPH AXIS
Growth hormone is secreted by somatotropes in the anterior pituitary gland. The secretory pattern is pulsatile, with discrete pulses of GH every 3 to 4 hours and virtually undetectable GH concentrations in between. Secretion of GH varies considerably with age114 and shows a sexually dimorphic pattern,115 with a greater average daily GH output in women. This pattern is the result of an interaction between the hypothalamic peptides GHRH and somatostatin (SS). The amplitude of the GH peak is determined by GHRH that stimulates the pituitary somatotrophs to increase both the secretion of stored GH and GH gene transcription. SS determines trough levels of GH by inhibition of GHRH release from the hypothalamus and GH release from the pituitary. Withdrawal of SS, on the other hand, determines the timing of a GH pulse.
More recently, the use of synthetic GH-releasing peptides (GHRP) has led to the identification of a GH secretagogue (GHS) receptor (GHS-R type 1a). The receptor is strongly expressed in the hypothalamus, but specific binding sites for GHRP have also been identified in other regions of the CNS and peripheral endocrine and nonendocrine tissues in both humans and other organisms.116,117 The endogenous ligand for the GHS receptor, ghrelin, was isolated from the stomach and is an octynylated peptide consisting of 28 amino acids.118 It is expressed predominantly in the stomach, but lower amounts are present within the bowel, pancreas, kidney, immune system, placenta, pituitary, testis, ovary, and hypothalamus.119 Ghrelin leads not only to the secretion of GH but also stimulates prolactin and ACTH secretion. Additionally, it influences endocrine pancreatic function and glucose metabolism, gonadal function, appetite, and behavior. It can also control gastric motility and acid secretion and has cardiovascular and antiproliferative effects. The role of endogenous ghrelin in normal growth during childhood remains unclear. Both ghrelin and GHRPs release GH synergistically with GHRH.
The expression of the human GH gene is regulated not only by a proximal promoter but also by a locus control region (LCR) 15 to 32 kb upstream of the GH-1 gene. The LCR confers pituitary-specific, high level expression of GH.120,121 The full-length transcript from the GH1 gene encodes a 191-amino-acid, 22-kD protein that accounts for 85% to 90% of circulating GH. Alternative splicing of the mRNA transcript generates a 20 kD form of GH that accounts for the remaining 10% to 15%. Within both the proximal promoter and the LCR are located binding sites for the pituitary-specific transcription factor Pit1. Additional binding sites for the transcription factor Zn15 are also located within the proximal promoter.
In the circulation, GH binds to two binding proteins, high-affinity GHBP and low-affinity GHBP.122 Little is known about the low-affinity GHBP, which accounts for approximately 10% to 15% of GH binding, with a preference for binding to 20 kD hGH. The high-affinity GHBP is a 61-kD, glycosylated protein that represents a soluble form of the extracellular domain of the GH receptor that can bind to both 20 and 22 kD hGH and thereby prolong the half-life of GH. In-vivo studies that have co-administered GH and GHBP to hypophysectomized and GH-deficient rats have demonstrated a potentiation of weight gain and bone growth, although similar studies have not as yet been performed in man.123
The GH receptor (GHR) is present in a number of tissues. The hormone sequentially dimerizes its receptor, activating a receptor-associated tyrosine kinase JAK2 that in turn is auto-phosphorylated and also phosphorylates the GHR. This then leads to signal transduction using the MAPK, STAT and PI3 kinase pathways. The end result is activation of a number of genes that mediate the effects of GH. These include early-response genes encoding transcription factors such as c-jun, c-fos, and c-myc—implicated in cell growth, proliferation, and differentiation—and IGF-I that mediates the growth-promoting effects of GH.124,125
IGF-I and IGF-II are single-chain polypeptide hormones that are widely expressed. Together with a family of specific binding proteins, they are believed to mediate most of the actions of GH.
NEONATAL PRESENTATION
Recent studies in humans and in animal models have demonstrated marked similarities but also critical differences between the clinical features of GHD and various forms of IGF deficiency.126–128 In GHD, prenatal growth is near normal, although mild reductions in birth length and weight have been observed. GHD does not cause severe IUGR, whereas loss of placental GH does.129 However, loss of IGF-I in utero results in severe intrauterine growth restriction in both humans and mice,130,131 suggesting that IGF-I and the IGF-I receptor are critically involved in intrauterine growth. IGF-I synthesis and secretion in utero are not regulated primarily by pituitary GH.
IGF-I production comes under GH regulation either in the last few months of fetal life or shortly after birth and is well established by 6 months of age. Growth failure is greater for skeletal growth than for body weight, so infants and young children have an appearance of relative adiposity. Neonates may present with hypoglycemia, and this suggests the possibility of other pituitary hormone deficiencies, especially ACTH. Normoglycemia is only maintained when cortisol replacement therapy is commenced, suggesting that ACTH (and consequently cortisol) secretion is critical for glucose homeostasis. However, the GH-IGF-I axis also plays a role in maintaining glucose homeostasis, although IGHD is rarely associated with neonatal hypoglycemia. A diagnostic fast may be required to dissect CPHD from other causes of hypoglycemia, although the distinguishing feature from hyperinsulinism is the absence of ketone body formation in the latter.
The presence of concomitant gonadotropin deficiency is suggested by the presence of microphallus, cryptorchidism, and scrotal hypoplasia. Genital ambiguity would not be expected, owing to placental production of hCG. Prolonged jaundice with conjugated hyperbilirubinemia and cholestasis may also be observed, typically in patients with CPHD. The relative contributions of GH, ACTH, and TSH deficiency to this presentation are unclear. It is imperative that the diagnosis of pituitary insufficiency be considered in any infant (especially term) with hypoglycemia, cryptorchidism, and microphallus, or conjugated hyperbilirubinemia. Associated features that might indicate more widespread problems (midline defects of the face, a single central incisor, nystagmus, and/or optic nerve hypoplasia) should be looked for and MRI undertaken.
INFANT AND CHILDHOOD YEARS
After the perinatal period, the defining feature of GHD is growth failure. Reduced skeletal growth may be observed during the first 6 months of life in congenital GHD, but by 6 to 12 months of age, early growth failure is almost inevitable.132–134 Height velocity is usually between −2 and −5 standard deviations (SD) from the mean, leading to progressive height centile crossing. In patients with acquired GHD, the critical feature is a change in growth rate. Between the age of 2 years and the onset of puberty, children maintain their height percentile with remarkable integrity. Deviation from this channel (either acceleration or deceleration) needs investigation. Thus a child who has been growing along the 75th percentile but moves across to the 25th percentile warrants evaluation, even though his/her height may still be within the normal range.
Bone age is often delayed in patients with GHD, but this may not be so in acquired GHD. The close proximity of time to the growth failure or acquired GHD accompanied by accelerated puberty is occasionally seen in patients with intracranial tumors, when bone age may be accelerated.93,94 Delayed dentition may be observed, but in the absence of midline craniofacial abnormalities is otherwise normal. Other skeletal appearances include hypoplasia of facial bones, hypoplastic nasal bridge, frontal bossing, and delayed closure of sutures. Head circumference is usually at the lower limits of normal, indicating normal brain growth.
An increase in adiposity, particularly central adiposity, can be detected by careful measurement of skinfold thicknesses. Genital growth prior to the onset of puberty is usually proportional to body size. Puberty may be delayed, but in the absence of other endocrine deficiencies is otherwise normal.
Limited data are available on the adult height of untreated GHD patients. These results are often difficult to interpret because of (1) heterogeneity in the timing of GHD, (2) heterogeneity in the severity of GHD, (3) the presence or absence of other pituitary deficiencies, and (4) delay in puberty, resulting in late epiphyseal fusion. Wit et al.135 summarized the results from studies of 22 untreated men and 14 untreated women with severe isolated GHD and reported a mean adult height of −4.7 SD. In patients with untreated autosomal recessive GHD, Rimoin and colleagues reported mean adult heights of −7.4 SD.136 In patients with CPHD, adult height is often not as severely affected as in IGHD, presumably reflecting pubertal delay and late epiphyseal fusion.137
Diagnosis of Growth Hormone Deficiency in Childhood
The diagnostic evaluation of children with growth failure is complex because there are multiple causes for short stature (Table 26-4). In the pursuit of the diagnosis of GHD, other causes for short stature need to be considered and excluded. This is because the diagnosis of GHD is one of exclusion.138 GH is the final common pathway for postnatal growth, and many causes of poor growth may secondarily affect GH secretion.139 There are a number of tests available for assessing GH status. Considerable attention has been paid to the underlying mechanisms assessed by the tests, how the samples should be collected, and what type of measurement should be performed. Less attention has been paid to the statistical assumptions underlying the performance of diagnostic tests. The statistical theory behind many tests is complex because the results do not follow an all-or-none law. Rather than being left with a clear-cut answer to the initial diagnostic question, the clinician is more likely to be left with a series of probabilities as to whether or not the patient is likely to have GHD.
Table 26-4. Causes of Short Stature
| Nonpathogenic |
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|