FIGURE 80-1. Portrait of Dr. Robert J. Graves.
(From Taylor S: Robert Graves. The Golden Years of Irish Medicine. New York: Royal Society of Medicine Services, 1989.)
Epidemiology
Graves’ disease is a relatively prevalent disorder, and it is the most frequent cause of thyrotoxicosis in iodine-sufficient countries.14
Several studies have attempted estimating the exact frequency of Graves’ disease in the general population. However, comparison of surveys is difficult because of the use of different criteria in population sampling, because of ethnic differences, and because diagnostic tools have changed over the years. In the United States, a large survey performed in the 1970s estimated the prevalence of Graves’ disease to be 0.4%.15 A similar prevalence (0.6%) was found in the Pescopagano study in Italy.16 The Whickham survey in the United Kingdom suggested a prevalence of 1.1% to 1.6% (i.e., about threefold to fourfold higher) for thyrotoxicosis of all causes, of which Graves’ disease was presumably the most frequent.17,18 A recent study performed in Sweden has shown an incidence of Graves’ disease of approximately 25 cases/100,000 per year, which is relatively high compared with other studies, a possible interpretation of which is a high iodine intake of the selected population (see later discussion).19 Overall, a meta-analysis of various studies has estimated the general prevalence of the disorder to be about 1%,20 which makes it one of the most frequent clinically relevant autoimmune disorders.
The dietary iodine supply appears to be a major factor in determining the frequency of Graves’ disease.21–28 For example, iodine supplementation of previously iodine-deficient Tasmania induced a threefold increase in the incidence of hyperthyroidism in 3 years.21 Although the increase was mainly due to iodine-induced thyrotoxicosis in patients with autonomously functioning nodules or goiters, it was also shown that LATSs or LATS protectors were present in a number of cases of thyrotoxicosis in the early supplementation period, thus suggesting that some of the iodine-induced cases of hyperthyroidism might have been due to Graves’ disease. Since the Tasmanian report, outbursts of iodine-induced thyrotoxicosis have been reported in many countries after the implementation of iodine supplementation programs.22–28 Thyrotoxicosis occurred primarily in older people with preexisting nodular goiter. However, in a study performed in Switzerland, a slight and transient increase in the incidence of Graves’ disease was noted after stepwise, full iodine supplementation (Fig. 80-2).23 Similar increases in the incidence of thyrotoxicosis have been reported in Sweden (16.6/100,000),24 New Zealand (15/100,000),25 Britain (23/100,000),26 and Denmark27, in the latter population especially in younger age groups.28 Population-based studies also show differences in the incidence of Graves’ disease in populations with different but relatively constant iodine intake. Nevertheless, this corresponds to a higher frequency of nonautoimmune thyrotoxicosis in iodine-deficient areas. Thus, by comparing two genetically similar populations that differed in terms of iodine intake (iodine-sufficient Iceland versus iodine-deficient East Jutland in Denmark), it was found that the incidence of Graves’ hyperthyroidism was slightly higher in the iodine-sufficient (20/100,000 inhabitants/year) than in the iodine-deficient population (15/100,000/year), but that the incidence of thyrotoxicosis of all causes was greater in the iodine-deficient (39/100,000/year) than in the iodine-sufficient (23/100,000/year) population.14 Clearly, these findings show that fear of an increased incidence of Graves’ disease should not prevent the implementation of iodine supplementation programs.
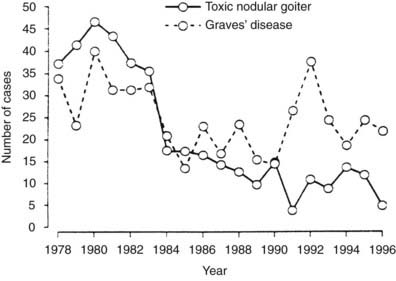
FIGURE 80-2. Increased incidence of hyperthyroidism from Graves’ disease and toxic nodular goiter in Switzerland after the introduction of iodine supplementation.
(Modified from Burgi H, Kohler M, Morselli B: Thyrotoxicosis incidence in Switzerland and benefit of improved iodine supply. Lancet 352:1034, 1998.)
Although ethnic differences in susceptibility to Graves’ disease are likely to exist, they have not been consistently investigated in comparative studies. As with many other autoimmune disorders, Graves’ disease is about fivefold more prevalent in women than men. The reasons for this observation are understood only in part, but some hypotheses will be discussed later. The annual incidence is clearly and consistently related to age, with peaks in the fourth to sixth decades of life,28 although Graves’ disease can be observed in people of any age, including children.
Etiology
Most of the pathogenic mechanisms of Graves’ disease have been clarified since the first description of LATS.11 It is now well established that Graves’ disease is an organ-specific autoimmune disorder, with involvement of both T- and B-cell-mediated immunity against thyroid antigens. TSHR is the main antigen involved, and circulating autoantibodies against TSHR (TSH receptor antibodies [TRAbs]) that are capable of stimulating the receptor are responsible for the most distinctive features of the disease, namely hyperthyroidism and goiter. Nevertheless, in spite of advancements in understanding the pathogenic mechanisms of Graves’ disease, the ultimate cause of the disease remains elusive. The majority of investigators share the opinion that Graves’ disease is a multifactorial disease caused by a complex interplay of genetic, hormonal, and environmental influences that lead to the loss of immune tolerance to thyroid antigens and to the initiation of a sustained autoimmune reaction.
GENETICS OF GRAVES’ DISEASE
It is common for endocrinologists to observe familial clustering of Graves’ disease by simply eliciting the family history of patients. Besides this common knowledge, a body of evidence indicates the existence of a genetic predisposition to Graves’ disease.
The strongest data in support of a genetic predisposition to Graves’ disease comes from twin studies.29,30 Dizygotic twins share on average 50% of their genome, whereas monozygotic twins share 100%. Moreover, twins are likely to share environmental factors more than any other kind of siblings. Several large twin studies have reported greater concordance rate of Graves’ disease in monozygotic than in dizygotic twins.29 Data obtained with modern diagnostic tools have shown a relatively low (~17% to 35%), but still significant, concordance in monozygotic twins.29,30 These findings clearly show a genetic influence, possibly characterized by low penetrance of the genes involved.
Another tool widely used to establish the existence of a genetic predisposition to any condition is family studies, in which the prevalence of the disease in relatives of index cases is compared with the prevalence in the general population. Early family studies showed a high prevalence of Graves’ disease and other thyroid abnormalities in first-degree relatives of patients with Graves’ disease and Hashimoto’s thyroiditis.31,32 The prevalence of circulating thyroid autoantibodies in siblings of patients was as high as 56% in some studies,31 which suggested a dominant mode of inheritance. These observations have been consistently confirmed in highly selected populations,33 but the results may not be applicable to the general population because of ascertainment bias. In an extensive segregation analysis with randomly ascertained probands, circulating antibodies were found in only 25% of the offspring of positive parents and in 14% of the offspring of negative parents, thus suggesting a multigenic model with less than 100% penetrance for the antibody trait.34 With the exception of very early studies, the prevalence of overt Graves’ disease has been found to be relatively low in siblings of patients.33,36 However, initial abnormalities in thyroid function compatible with subclinical hyperthyroidism or hypothyroidism have been reported.33,35 Villanueva et al. found that 36% of Graves’ patients with ophthalmopathy have a family history of either Graves’ disease or autoimmune thyroiditis, which in 23% of the cases affected first-degree relatives.37 Autoimmune thyroiditis is frequently observed in siblings of probands with Graves’ disease, as well as the contrary,33,37 suggesting that the two diseases may share some susceptibility genes that predispose to thyroid autoimmunity, but that the full expression of the phenotype depends on other genes and/or on environmental factors. Other organ-specific, non-thyroid-related autoimmune diseases may also be more prevalent in relatives of patients with Graves’ disease.33
These data and those obtained in twin studies are indicative of a complex multigenic pattern of inheritance of Graves’ disease. Some of the components of the phenotype, such as the presence of circulating antithyroglobulin and antithyroperoxidase antibodies, may be inherited in a dominant fashion with high penetrance.33 However, these genetic determinants do not appear to be sufficient for full expression of the disease. Clearly, other genes must be involved, which is in line with the complexity of the inheritance observed. Also, it appears from epidemiologic and experimental data that environmental factors (reviewed later in this chapter) play an important role by modulating the effect of an inherited predisposition. Based on the above evidence, a number of genes or loci have been investigated as candidates for predisposing factors (Table 80-2).33,38
Table 80-2. Genetic Determinants Associated With Graves’ Disease
| Gene | Possible Mechanism | Evidence in Favor of an Association |
|---|---|---|
| HLA-DR | Altered antigen presentation | Fair |
| CD40 | Altered antigen presentation | Fair |
| CTLA-4 | Altered antigen presentation | Good |
| PTPN22 | Altered T-cell activation | Fair |
| Thyroglobulin | Loss of tolerance | Good |
| TSH-R | Loss of tolerance | Poor |
Genes Predisposing to Graves’ Disease
In the last 20 years, the impressive advancement of biomedical research has allowed a remarkable expansion in genetics, which has led to the identification of several genes involved in the predisposition to Graves’ disease.33,38 Linkage, association, and candidate genes analyses, as well as whole-genome screening, have all been used to accomplish this goal. Based on the data available, genetic susceptibility to thyroid autoimmunity in general and to Graves’ disease in particular seems to be determined by a combination of a number of genes, some which most likely still remain to be identified.33,38,39
1: The HLA Complex
The HLA complex, which is located on the short arm of human chromosome 6, contains the sequence encoding about a hundred genes, most of which are involved in regulation of the immune response.40–42 The HLA genes are classically grouped into three major classes. Class I includes histocompatibility genes expressed on the surface of most cells (HLA-A, HLA-B, and HLA-C). Class II includes histocompatibility genes expressed exclusively on the surface of leukocytes and immune cells (HLA-DR). Class III includes a heterogeneous group of genes encoding molecules involved in the immune response, such as some complement factors, cytokines, and lymphocyte surface molecules. Other genes in this class are not clearly related with immunity. Most genes of the HLA complex are highly polymorphic, which makes them excellent candidates for disease susceptibility.
Experimental thyroiditis in the mouse was the first autoimmune disease to be associated with HLA.43,44 Early population studies in humans indicated an association of Graves’ disease with HLA-B8 and a relative risk of 3.9 in white patients.45 Subsequent studies also suggested an influence of that haplotype on the clinical course of the disease.33,44 However, HLA-DR3 (HLA-DRB1*03) was later shown to increase the risk to a greater extent and was considered to be the true determinant of the disease because it was in linkage disequilibrium with the B8 allele.33 Among Caucasians, HLA-DQA1*0501 was found to confer a relatively high risk within DR3 itself.33,44 This allele is in linkage disequilibrium with both B8 and DR3 and gives a relative risk of 3 to 4 for Graves’ disease in the white population. Sequencing of the DRβ-1 chain of HLA-DR3 allowed the identification of Arg74 as the critical amino acid conferring susceptibility to Graves’ disease.33,44
Different haplotypes seem to be involved in ethnic groups other than Caucasians: DQ3 in patients of African descent and Bw46 in those of Asian descent, although the data available are limited and have not always been reproducible.33,44
In general, HLA associations have been shown to confer a relatively low risk, even with alleles that have a high prevalence in the general population. Thus linkage analysis, a powerful tool for mapping essential predisposition genes, has been negative when the HLA region was examined using different polymorphisms at the same locus.33,44 Overall, it seems that the HLA locus explains a small fraction of the total genetic predisposition, but it is neither the major nor the only determinant, although it represents an established risk-increasing factor.
2: CD40
CD40, a member of the tumor necrosis factor receptor family, is expressed in B cells and other antigen-presenting cells and is involved in B-cell activation and proliferation, antibody secretion, immunoglobulin class switching, affinity maturation and generation of memory cells.46 Linkage studies have shown an association of the CD40 gene with Graves’ disease and the subsequent sequencing of the gene led to the identification of a C/T polymorphism at the 5′ untranslated region of CD40 strongly associated with Graves’ disease.33 This polymorphism influences the translational efficiency of the gene, which may have functional consequences in the CD40 protein.
3: CTLA-4
CTLA-4 is a T-lymphocyte surface protein with a major role in down-regulation of the immune response.47 Several studies have provided evidence that CTLA-4 is linked to Graves’ disease, autoimmune thyroiditis, and to the production of autoantibodies against thyroid antigens.33,44–48 Several variants of the CTLA-4 gene have been implicated in its possible causative role in Graves’ disease, among which a CTLA-4 polymorphism at position 60 was found to be the most suitable candidate in a large comprehensive analysis, the functional relevance of the polymorphism being possibly due to a reduced mRNA expression encoding the soluble form of the molecule.47 Nevertheless, a subsequent study did not confirm these data.48 Although CTLA-4 seems to be a genetic determinant of Graves’ disease, the causative variant remains to be identified with certainty, and it is possible that a haplotype consisting of more than one variant is responsible for the association.33
4: The Protein Tyrosine Phosphatase 22 (PTPN22) Gene
PTPN22 is a powerful inhibitor of T-cell activation.49 A single nucleotide polymorphism at codon 620 associated with other autoimmune diseases was found to be associated with both Graves’ disease and autoimmune thyroiditis, with significant ethnic differences in the association.49,50
5: Thyroglobulin
Thyroglobulin is the precursor of thyroid hormones and a major autoantigen in thyroid autoimmunity. Recent wide genome screens have provided evidence for a strong linkage between a locus on chromosome 8q24, where the thyroglobulin gene is located, and autoimmune thyroid diseases.33,51 Sequence analysis of the thyroglobulin gene has shown numerous single nucleotide missense polymorphism, suggesting that amino acid variants in the Tg proteins may contribute the pathogenesis of autoimmune thyroid diseases, including Graves’ disease.33,52
6: TSH-R
Despite the central role of TSH-R in the pathogenesis of Graves’ disease, the association of the disease with the gene encoding the receptor remains controversial.33 Although three common missense single nucleotide polymorphisms have been found to be associated with Graves’ disease, these associations were not always confirmed by other studies. The extent of the contribution of the TSH-R gene remains to be established.33
7: Other Genes
In the search for genetic determinants of Graves’ disease, several other genes have been studied over the years, namely genes involved in the immune response. The immunoglobulin genes were studied extensively, but conflicting results were observed in association studies.33 Other candidate immunoregulatory genes that have been studied include interleukin 1 (IL-1), IL-1 receptor antagonist, tumor necrosis factor receptor 2 (TNF-2), and interferon γ (INF-γ). None of these genes showed significant associations with Graves’ disease.33 Additional loci have been linked in families with Graves’ disease, including one on chromosome 14q31 (GD-1), one on chromosome 20 (GD-2), and one on chromosome Xq21-22 (GD-3).33
ENVIRONMENTAL FACTORS AND GRAVES’ DISEASE
The relative low penetrance in twins and first-degree relatives of patients with Graves’ disease suggests that environmental factors must play a major role in inducing the disease in genetically susceptible individuals.33,53,54 Several studies have shown that various nongenetic factors may in fact contribute to the development of Graves’ disease.
Infections
Over the years, both experimental and epidemiologic evidence has suggested that infections could play a role in the pathogenesis of Graves’ disease.54,55 Seasonal and geographic variations in the incidence of the disease have been reported,56,57 although seasonal variations have not been confirmed in other studies.58 Blood group nonsecretors, who are more prone than secretors to infections, are more frequently found among patients with Graves’ disease than in controls.59 This observation has been interpreted as indirect evidence that infectious pathogens may be involved in the etiology of Graves’ disease, although a direct genetic effect of the secretor status could also explain these results. Evidence of a recent viral infection has been reported in a high percentage of patients with Graves’ disease.54,55
Molecular mimicry has been invoked to explain the association between infections and Graves’ disease.60 Molecular mimicry is based on the hypothesis that cross-reactions of some microbial antigens with a self-antigen may cause an immune response to autoantigens. In Graves’ disease, the pathogen Yersinia enterocolitica has been thoroughly studied after reports of association of this microbe with the disease. A high prevalence of circulating antibodies against Y. enterocolitica has been observed in patients with Graves’ disease, and Yersinia antibodies were found to interact with thyroid structures.61–63 In a recent study from Denmark, it was shown that the occurrence of IgA and IgG antibodies against Yersinia not only is more frequent in Graves’ patients than in case controls but also in twins with Graves’ disease compared with their discordant twins.64 Saturable binding sites for TSH have been found in Yersinia and were also recognized by TRAbs from patients with Graves’ disease.65,66 In animals immunized with Yersinia proteins, antibodies developed against human thyroid epithelial cells and TSHR.67,68 Overall, the affinity of these cross-reactive antibodies to the thyroid was low, and immune responses were transient. Low-affinity binding sites for TSH have also been found in other bacteria, including some species of Leishmania and Mycoplasma.53,54 However, it must be noted that thyroid autoimmunity does not develop in most patients with Yersinia infections,69 so the evidence in favor of Yersinia infections as a precipitating cause of Graves’ disease awaits confirmation.
Viruses could theoretically trigger autoimmunity through several mechanisms, including interactions with autoantigens, permanent expression of viral proteins on the surface of epithelial cells, aberrant induction of HLA antigens on epithelial cells (see later), and molecular mimicry.60,70 In 1989, the presence of retroviral (HIV-1 glycosaminoglycan protein) sequences in the thyroid and peripheral mononuclear cells of patients with Graves’ disease was reported,71 but viral sequences were not found in control thyroids. This finding, however, remained isolated and was not confirmed in subsequent studies.72,73 Human foamy virus antigens were shown by immunofluorescence to be present in the thyroid of patients with Graves’ disease.74 Again, further studies using more specific and sensitive techniques failed to identify foamy virus DNA and antiviral antibodies in the blood of affected subjects.75,76 Homology between another HIV-1 protein (Nef) and human TSHR has also been reported, although sera from patients with Graves’ disease did not react with the peptide bearing the highest degree of homology.77 Another retroviral protein, p15E, has been isolated from the thyroids of patients with Graves’ disease but not from control glands.78 In this regard, it is worthwhile emphasizing that retroviral-like proteins, including p15E, are encoded by the normal human genome. Although their function is unclear, they may be expressed in many epithelial tissues under certain conditions such as inflammation and may modulate but not initiate the immune response.79 The finding of retroviral sequences or proteins in the glands of patients with Graves’ disease may therefore represent a secondary rather than a causative phenomenon. Circulating antibodies against another retroviral particle, namely HIAP-1, have been found in as many as 87.5% of patients with Graves’ disease as compared with 10% to 15% of controls,80 but HIAP-1 particles were not detected when human T cells were co-cultured with Graves’ thyrocytes.81
A highly speculative hypothesis has been raised that involves superantigens.55,60 Superantigens are endogenous or exogenous proteins, such as microbial proteins, capable of stimulating a strong immune response through molecular interactions with nonvariant parts of the T-cell receptor and the HLA class II proteins. Through this mechanism, superantigens are in theory capable of stimulating the expansion of autoreactive T cells and therefore of driving an autoimmune response.55,60 Such a mechanism has been suggested in rheumatoid arthritis, and a similar mechanism was proposed for Graves’ disease. In vitro superantigen stimulation of glands with autoimmune thyroid disease induced expression of HLA class II molecules on thyrocytes, and this phenomenon was IFN-γ dependent.82 The interpretation of this observation was that superantigen-reactive T cells exist among the lymphocytes infiltrating the thyroid in autoimmune thyroid disease, and that these lymphocytes may have been activated after exposure to extrinsic superantigens.
The most recent hypothesis, the so-called “hygiene hypothesis of autoimmunity,” implies that infections may protect from, rather than precipitate, autoimmune diseases. Exposure of the immune system to infective agents may somehow allow better control of autoimmune responses. In this regard, improved living standards have been associated with decreased exposure to infections and an increased risk of autoimmune diseases. Kondrashova et al. reported a much reduced prevalence of thyroid autoantibodies in the lower-economic population, which may suggest the hygiene hypothesis may apply to thyroid autoimmune diseases.83 Further studies are needed to investigate whether this is in fact the case.
In summary, although epidemiologic evidence indicates that infection may play an important role as a causative or protective factor in Graves’ disease, we are still lacking definitive identification of the etiologic organism(s) and a reasonable explanation for microbes to precipitate or protect from the disorder.
Stress
The suggestion that psychological stress may be a precipitating factor in Graves’ disease has been made as early as the first description of the disease.3 The occurrence of stressful events before the onset of Graves’ disease is a recurrent impression among clinicians, and by the end of the 19th century, Graves’ disease was considered to be a result of prolonged emotional disturbances. In cross-sectional questionnaire-based studies, some investigators have shown an increased prevalence of stressful life events in the months preceding the onset of Graves’ disease.84–87 However, some of the recorded events (such as arguments with spouses and in the workplace) could have been influenced by the behavior of patients with a yet undetected hyperthyroidism and therefore be a consequence rather than a cause of the disease. Other events, however, were largely independent of the patient’s behavior, such as unemployment and financial difficulties. In some studies, patients were asked to rate the stressfulness of life events, and patients with Graves’ disease ranked such events more stressful than did controls.84 Thus it is possible that the perception of life events is different in hyperthyroid patients. In cross-sectional studies, an increase in the prevalence of Graves’ disease was reported during World War II in Germany but not during the civil unrest in Ireland or during the German occupation of Belgium, which suggests that the stressful events of personal life may be more important than “social stress.”84 On the other hand, chronic stress due to panic disorder was not associated with the occurrence of Graves’ disease.88 Stress is associated with increased adrenocorticotropic hormone (ACTH) and cortisol secretion, which can in turn determine immune suppression, but additional non-ACTH-related immunosuppressive phenomena also occur.89,90 Recovery from such immune suppression can be associated with rebound immune hyperactivity, which could precipitate autoimmunity. The best example of such a phenomenon is perhaps the well-documented immune suppression of pregnancy, which can be followed by new or recurrent onset of autoimmune disorders, including Graves’ disease (see later discussion).91
In summary, limited but significant evidence indicates that stress may be a contributing factor in the etiology of Graves’ disease, probably in connection with other predisposing factors. The available studies are all retrospective and therefore carry a number of possible biases. Worried patients sick with hyperthyroidism may be more prone to recall upsetting events, and subclinical undetected thyrotoxicosis present before clinical diagnosis may alter the perception of life events and even the behavior of patients. Unfortunately, prospective studies addressing the problem of stress and Graves’ disease and its interplay with genetic factors are not available to date.
Gender
Graves’ disease is typically but not exclusively a disease of women. In most series, the female-to-male ratio ranges from 5 to 10 at any age,16,17 although the difference may be smaller during childhood.92 The reason for the disproportionate prevalence of Graves’ disease in women is not known, but genetic and nongenetic factors must play a role. A number of studies indicate a stronger immune system in women.93,94 Autoimmune phenomena and diseases are in general more prevalent in women.93,94 A large body of evidence clearly indicates the existence of sexual dimorphism in normal and abnormal immune responses in spontaneous and experimental animal models, including models of autoimmune thyroiditis.93,94 In these models, male hormones appear to down-regulate immunity and therefore protect animals from autoimmunity, whereas the effect of estrogen is not always unequivocal. Despite the evidence obtained from animal studies, little evidence in the literature supports a role for sex hormones in the high prevalence of Graves’ disease in women.93,94 Women with normal baseline levels of estrogen, but with an increased sensitivity to the hormone as shown by the presence of melasma, had a higher prevalence of thyroid autoimmune disorders.95 However, a clear association between exogenous estrogen administration and Graves’ disease has never been reported. Moreover, thyroid autoimmunity is often found in patients with Turner’s syndrome, who typically have low estrogen levels.96 Conversely, male patients with primary hypogonadism such as in Klinefelter’s syndrome do not show a higher incidence of Graves’ disease or Hashimoto’s thyroiditis.97 In these human conditions, however, the chromosomal abnormality probably plays an important role that is largely independent of sex hormone levels.
Pregnancy is an important risk factor, and it is well recognized that in any woman the risk of development of Graves’ disease increases fourfold to eightfold in the postpartum year.91 The abrupt fall in the level of pregnancy-associated immunosuppressive factors immediately after delivery (rebound immunity) is likely to be the mechanism responsible for the precipitation of Graves’ disease.91 Nevertheless, in a recent retrospective study it was found that the relative frequencies of postpartum onset of Graves’ disease were similar in relation of increasing parity, which would not support a role of the postpartum period as a major risk factor for the first appearance of Graves’ disease.98 On the other hand, it was also shown by the same group that the postpartum period is indeed a risk factor for relapse of Graves’ thyrotoxicosis after withdrawal of antithyroid drugs.99 The factors involved in the immune alterations of the postpartum period may include but are not limited to estrogen and progestin.91,100
Besides sex hormones, factors on the X chromosome could explain the epidemiologic evidence of a female preponderance in Graves’ disease. A linkage analysis in families with Graves’ disease has located a putative Graves’ disease susceptibility locus on the long arm of the X chromosome.33 Although most X-linked disorders are expressed phenotypically only in men, it is possible that a gene with a dose-dependent effect may determine more relevant clinical effects in women. This finding could help explain the higher incidence of Graves’ disease observed in women and, possibly, in patients with Turner’s syndrome. Inactivation of the X chromosome, an epigenetic phenomenon, has also been suggested to be involved in the female predisposition to thyroid autoimmunity, and recently it was shown that this phenomenon is more frequently observed in patients with Graves’ disease or autoimmune thyroiditis than in healthy controls.101
Smoking
A number of studies have provided evidence for an association between smoking and thyroid diseases, including Graves’ disease.102,103 Retrospective analysis shows that in smokers there is an increased risk of Graves’ disease and ophthalmopathy, as well as of relapse of hyperthyroidism following anti-thyroid drug withdrawal, which is more pronounced in the female gender. The findings may be explained both by a direct action of smoking metabolites on the immune system or by damage induced by smoking metabolites on thyrocytes, which may determine exposure of thyroid antigens to the immune system.
Thyroid Damage
There are reports of Graves’ disease appearing after ethanol injections performed for treatment of autonomous thyroid nodules, which has been interpreted as due to the massive release of thyroid antigens, thereby triggering an autoimmune response to TSH-R in predisposed individuals.104 Furthermore, and possibly because of similar pathogenetic mechanisms (i.e., massive release of thyroid antigens), Graves’ disease or simply serum TRAb have been reported to appear following radioiodine treatment for toxic adenoma or toxic nodular goiter.105 We observed a patient with Graves’ disease that appeared after a neck injury (unpublished observation), which in theory may also reflect release of thyroid antigens.
Pathology
It is now rare to observe the full pathologic changes that occur in the thyroid glands of untreated patients.106 On gross pathology the gland is significantly enlarged, with a smooth and hyperemic surface. A prominent pyramidal lobe is often visible, and the contour of the gland is irregular with multiple lobulations. Microscopically, both hypertrophy and hyperplasia are found. Follicles are small, with scanty colloid as a result of ongoing thyroid hormone secretion. The follicular epithelium presents a columnar aspect, with even a pseudopapillary appearance. Blood vessels are large and congested. Various degrees of lymphocytic infiltration can be found between the follicles. T cells predominate in the interstitium, whereas B cells and plasma cells predominate in the occasional lymphoid follicles. On electron microscopy, the cellular hyperactivity is demonstrated by an increase in the Golgi reticulum and the number of mitochondria and by the presence of prominent microvilli. With longstanding Graves’ disease, distinct nodularities with an adenomatous appearance may develop, and the lymphocytic infiltrate may become more prominent and resemble chronic thyroiditis.
This pathologic picture of active Graves’ disease is dramatically changed by antithyroid drugs and iodine treatment, which is now universally performed before surgery. Vascularity and vascular congestion are much less pronounced, and the follicles can be larger.106
Pathogenesis of Graves’ Disease
Although the ultimate cause of Graves’ disease is still unknown, over the years a large body of evidence has accumulated and provided important insight into the immune mechanisms that eventually lead to the clinical manifestations of the disease. Since their first description in 1956,11 much attention has been devoted to the study of TRAbs. It has also been recognized that although TRAbs are the ultimate cause of both goiter and hyperthyroidism, the nature of the immune dysfunction involves many aspects of the immune system, including changes in both B-cell and T-cell function. The follicular cell per se may also play an independent role.
ROLE OF TSH RECEPTOR ANTIBODIES
Nomenclature
The nomenclature of TRAbs is complex and largely dependent on the assay used to detect these antibodies in serum.107–111 Assays measuring displacement of radiolabeled TSH from its receptor by serum immunoglobulins detect TRAbs regardless of their functional activity. These antibodies have been termed TSH-binding inhibitory immunoglobulins (TBIIs). Assays for TSAbs use cellular systems carrying a functional TSHR and detect the release of cyclic adenosine monophosphate (cAMP) in the culture medium upon challenge with serum or purified immunoglobulins.107–111 These antibodies are essentially the cause of hyperthyroidism in Graves’ disease. In the same bioassay system, antibodies with blocking activity on the TSHR (TBAbs) can be detected.107–112 These antibodies characterize a subset of patients with atrophic autoimmune thyroiditis and hypothyroidism, but they can also occasionally be found in patients with Graves’ disease, in combination with TSAbs.112
Major Autoantigen in Graves’ Disease: Structure-Function Relationship of the TSHR
Definitive proof that TSHR is the target of TSAbs came from the cloning of this protein in the late 1980s.12,13,107–109 The receptor is a member of the G protein–coupled receptor superfamily. Its structure includes seven hydrophobic transmembrane domains, an extracellular N-terminal domain (ectodomain), and an intracellular C-terminal domain. Heavy glycosylation of the extracellular domain accounts for about 20% of its molecular weight of 84 kD. The primary structure of the protein consists of 744 residues. The gene is located on chromosome 14q31 and is formed by 10 exons that yield a single polypeptide. Shedding of the N-terminal extracellular 310 amino-acid residues (the so-called A subunit) may either initiate or amplify the immune response against TSHR, being immunoreactive epitopes partially sterically hindered on the holoreceptor on the plasma membrane.108,113,114 Binding sites for TSH and TRAbs are located in the extracellular domain and are mainly conformational.107,108 The major epitopes for TSAbs are located in the extreme N-terminal portion, whereas those for TBABs are mainly, but not exclusively, in the C-terminal portion of the extracellular domain, closer to the cell membrane.107,108,115–118 However, this should not be taken as a paradigm, especially since TBABs are more diverse and spread over the receptor extracellular domain.108 In fact, TRAbs are to some extent heterogeneous for epitope recognition, possibly due to epitope spreading during the immune response.108 Whether and to what extent glycosylation and dimerization of TSH receptor affects its recognition by antibodies is uncertain.108
Assays for TSH Receptor Antibody
The pioneering era of bioassays in vivo for TRAbs11,119 has been superseded by the present period, in which a number of in vitro assays are more readily performed by many laboratories and provide more reproducible and reliable measures of TRAb levels in the serum of patients. The radioreceptor assay uses TSHR from various sources: porcine or human recombinant TSHR from transfected cell lines.120–126 Although assay design varies in all these methods, they all rely on displacement of labeled TSH from solubilized TSHR from the serum of patients. Studies in hyperthyroid patients with Graves’ disease show positive TBII tests with these methods in 75% to 95% of patients. The recently designed second-generation radioreceptor assay attains even higher sensitivity while maintaining high specificity (99%)126 and is commonly used by the majority of laboratories for the routine detection of TRAb. An enzyme-linked assay is also available.127 Radioreceptor assays do not require permanent cell cultures; they are the most readily available (also commercially) and are therefore the most frequently used in clinical practice. The TBII test, however, does not give information on the functional properties of the antibody detected and can be positive in the presence of TBAb.
The functional stimulating properties of TRAbs can be studied by in vitro bioassays based on the measurement of cAMP production from cells with a functional TSHR.120 Human thyroid follicular cells,128 a rat thyroid cell strain (FRTL-5),129,130 and Chinese hamster ovary cells stably transfected with human TSHR (CHO-R)131–133 have all been used for this purpose. With these assays, TSAb can be detected in more than 90% of patients with untreated Graves’ disease (Fig. 80-3). The system using CHO-R cells has some advantages over the others: it is slightly more sensitive and relies on easier culture conditions, which also makes it more reproducible in different laboratories. As stated previously, the bioassays have the advantage of giving information on the functional properties of TRAbs and, in a modification of the assay, can also identify TBAbs.112,133 However, they require permanent cell culture equipment and pre-purification of the immunoglobulin fraction of serum, which makes these assays not readily available to routine endocrine laboratories. The latter problem has been overcome by very sensitive assays in which activation of a transfected firefly luciferase gene produces chemiluminescence in response to TSHR stimulation by whole serum.134,135 Even more recently, a coated-tube assay which discriminates TSAb from TBAb was developed.136 The assay is based on the use of a chimeric receptor, where a TSAb epitope 8-165 is replaced by comparable LH receptor residues. Binding of radiolabeled TSH to this chimera can be inhibited by sera containing TBAb up to 95%.
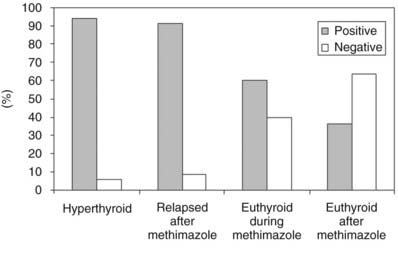
FIGURE 80-3. Prevalence of serum thyroid-stimulating antibody tests in Chinese hamster ovary cells transfected with recombinant human thyroid-stimulating hormone (TSH) receptor (CHO-R). Serum IgG was purified from untreated and treated patients with Graves’ disease.
(Modified from Vitti P, Elisei R, Tonacchera M et al: Detection of thyroid-stimulating antibody using Chinese hamster ovary cells transfected with cloned human thyrotropin receptor. J Clin Endocrinol Metab 76:499–503, 1993.)
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


