FIGURE 115-1. The primary amino acid sequences of naturally occurring decapeptide gonadotropin-releasing hormone (GnRH) structural variants spanning approximately 600 million years of evolution. Boxed regions show the conserved aminoterminal and carboxyterminal residues, which are involved in receptor binding and activation. The GnRHs are named according to the species in which they were first discovered, and they may be represented in more than one species. For example, mammalian GnRH present in amphibians and primitive bony fish, as well as chicken GnRH I (GnRH II), is present in most vertebrate species, including man. An octopus GnRH and an additional Ciona GnRH comprising 12 and 16 amino acids, respectively, but retaining the conserved aminoterminal and carboxyterminal domains are not shown.
(Data from Barran PE, Roeske RW, Pawson AJ, et al: Evolution of constrained GnRH ligand conformation and receptor selectivity, J Biol Chem 280:38569–38575, 2005.)
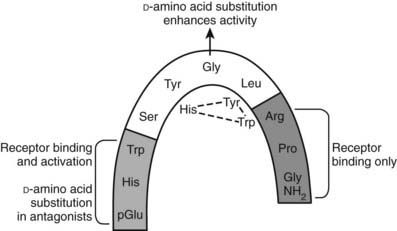
FIGURE 115-2. Schematic representation of gonadotropin-releasing hormone (GnRH) in the folded conformation in which it is bound to the GnRH receptor. The molecule is bent at glycine at position 6, where substitution with d-amino acids (as in GnRH agonist and antagonist analogs) stabilizes the folded configuration, increases binding affinity, and decreases metabolic clearance. The N and C termini are involved in receptor binding. The N terminus is involved in receptor activation, and substitutions in this region produce antagonists. The residues shown below the structure are the substitutions in GnRH II that may form a triad of interactions.
TERTIARY STRUCTURE OF GnRH
The notion that glycine in position 6 facilitates adoption of the βII′-type turn conformation of GnRH to promote binding to its receptor in vertebrates was supported by the demonstration that protochordate GnRHs with the chiral amino acid in position 6 had very poor activity at vertebrate GnRH receptors, but this activity could be enhanced more than 10-fold by substitution with the achiral glycine in position 6.69 In contrast, protochordate GnRH receptors did not distinguish between the GnRHs with different amino acid residues in this position, suggesting that the peptide is able to interact with receptor binding sites in a more extended form, and that the evolution of receptors requiring interaction with GnRH in the folded conformation only arose after the evolution of jawed vertebrates. Structural analysis of GnRHs with the use of a combination of ion mobility mass spectrometry and molecular modeling revealed a close correlation between the propensity for formation of the βII′-type turn conformation (e.g., mammalian GnRH and a Gly6-substituted protochordate GnRH) and higher binding affinities at vertebrate receptors.69 These findings indicated that the substitution of glycine for a chiral amino acid during evolution facilitates more constrained conformation of GnRH for receptor binding, and that this subtle single amino acid substitution at a site remote from the ligand functional domains has marked effects on its structure and activity.73–75
In the absence of a crystal structure of GnRH, indirect studies utilizing fluorescence spectroscopy,76 region-specific antisera,77 conformational memory approaches,78 and nuclear magnetic resonance (NMR)79,80 support the conclusion that mammalian GnRH I adopts the folded structure. A three-dimensional structure of GnRH has been described in a recent NMR report (PDB code: 1YY1).
GnRH ANALOGUES
Substitution of Gly6 with d-amino acids greatly enhances the propensity for GnRH to adopt the βII′-type folded conformation in which it engages with its cognate receptor.73,74,81,82 This greatly enhances the binding affinity and hence agonist potency. Moreover, this substitution conveys resistance to peptidases that cleave the Gly6-Leu7 bond. Substitution of d-amino acids with bulky hydrophobic side chains (e.g., D-Trp) also facilitates binding to plasma proteins, thus reducing renal clearance and increasing the half-life. Substitution of the carboxyterminal Gly-NH2 with N-ethylamide or azaglycine protects from carboxypeptidases and is incorporated into some agonists. The structures of GnRH therapeutic agonists with these substitutions are shown in Fig. 115-3.

FIGURE 115-3. Gonadotropin-releasing hormone (GnRH) agonist and antagonist analogues in clinical practice or in development.
(Data from Ref. 81.)
Because the NH2-terminal residues of GnRH have a crucial function in receptor activation, substitution of these residues with d-amino acids with bulky hydrophobic side chains in combination with d-amino acid substitutions of Gly6 and D-Ala-NH2 substitution of Gly10-NH2 create high-affinity (less than 1nM) and high-potency antagonists (see Figs. 115-2 and 115-3).
In spite of these improvements in properties, GnRH analogues retain a relatively rapid metabolic clearance and are not orally bioavailable. The rapid metabolic clearance has been overcome by formulation of long-acting injectable biodegradable polymers in which GnRH analogues are embedded or by production of analogues with slow-release microcrystalline or gelling properties. These preparations are active from 1 to 12 months after a single injection.
Nevertheless, benefits are derived from developing orally active small molecule antagonists, as these avoid the trauma of injection and allow rapid withdrawal from therapy, thereby also facilitating the possibility of intermittent therapy. They also provide the opportunity to adjust doses for individuals and partially suppress steroid hormones to avoid bone loss and other side effects associated with reducing steroids to castration levels. Several of these small molecule GnRH antagonists are currently being tested for use in man,66,83 and one is well advanced in clinical development.84
Clinical Applications
PULSATILE GnRH (STIMULATORY)
Natural GnRH administered in hourly pulses (to mimic hypothalamic secretion) is effective in treating hypogonadotropic hypogonadism (e.g., in anorexia nervosa, hypothalamic amenorrhea, and Kallman’s syndrome), undescended testes (cryptorchidism), and delayed puberty (Fig. 115-4).
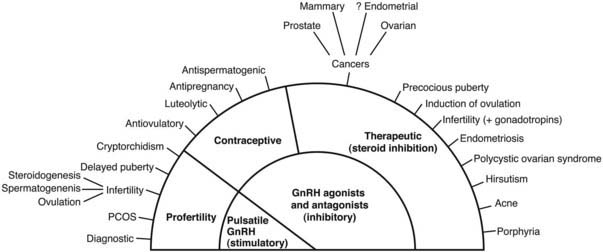
FIGURE 115-4. Clinical applications of gonadotropin-releasing hormone (GnRH) analogues.
(Data from Ref. 85.)
GnRH ANALOGUES (INHIBITORY)
GnRH analogues are used extensively as intervention therapies in a wide spectrum of pathologies, which is reflected in annual sales of about $2 billion. Long-term pharmacologic administration of GnRH agonists induces gonadotrope desensitization and inhibition of gonadotropin secretion, resulting in inhibition of gonadal sex steroid hormones and gametogenesis (see Fig. 115-4). This is a slow process, requiring 1 to 2 weeks, and induces stimulation before desensitization occurs. In contrast, GnRH antagonists have immediate inhibition of gonadotropins, with the rate of decline equal to that of their half-lives. Consequently, GnRH antagonists rapidly inhibit steroid hormone secretion. Therapeutic applications in hormone-dependent diseases include prostatic, breast, and ovarian cancer, endometriosis, uterine fibroids, precocious puberty, polycystic ovarian syndrome, and acute intermittent porphyria (see Fig. 115-4).27,85–91 GnRH agonists and antagonists are used extensively to inhibit endogenous gonadotropin, allowing regulated gonadotropin add-back for the induction of ovulation in in vitro fertilization (IVF) treatment of infertility. GnRH antagonists also hold promise as a new generation of male and female contraceptives with add-back gonadal steroids.83,92–94
OTHER ACTIONS OF GnRH
Although the primary function of GnRH is to regulate gonadotropins, it has become apparent during the ensuing 30 years since its discovery that the hormone and its receptors are also expressed, and have actions, in many other tissues. These include various regions of the preimplantation embryo, the placenta, endometrium, gonads, and central and peripheral nervous system, as well as many nonreproductive tissues such as the pancreas, eye, and olfactory epithelium in immune cells and in many cancer cells (see Reviews at references 1, 66, 81, 82, and 95–99). In view of these extensive reviews, the occurrence of GnRH/GnRH receptor and their actions will not be considered further here. Instead, we will focus on the effects of newly emerging GnRH actions on sexual behavior and inhibition of proliferation of cancer cells.
GnRH EFFECTS ON SEXUAL BEHAVIOR AND APPETITE
GnRH I and GnRH analogues and fragments of GnRH I were shown many years ago to stimulate lordosis in female rats.100 Subsequently, GnRH II was found to stimulate courting behavior in ring doves101 and to induce female solicitation of males in song sparrows.102 GnRH also stimulates spawning behavior in the goldfish.103 Another study demonstrated stimulation of male behavior by GnRH II treatment of female green iguanas.104 In the green anole,105 GnRH also stimulates reproductive behavior. These findings encouraged more detailed exploration of GnRH actions on reproductive behaviors in mammalian species. GnRH II stimulated sexual behavior in female musk shrews, and GnRH I was inactive.106 Moreover, an antagonist at the type I GnRH receptor, which is an agonist at the type II GnRH receptor (antagonist 135-18), stimulates reproductive behavior in female musk shrews, suggesting that the effect is mediated by the type II receptor.107 This conclusion is further supported by the demonstration that antide, which is an antagonist at the type I receptor but is inactive at type II receptors, had no effect on GnRH II stimulation of behavior. These effects could be demonstrated only after food restriction. It is interesting to note that GnRH II itself was found to inhibit feeding behavior in musk shrews.107,108 Thus, it appears that under normal conditions, when animals are satiated, endogenous GnRH II is maximally stimulating reproductive behaviors and exogenous GnRH II is therefore ineffective. In food-restricted animals GnRH II secretion in the midbrain appears to be decreased, such that exogenous GnRH II administration can reinstate reproductive behaviors. Therefore, there appears to be an interesting dual effect of GnRH II in stimulating reproductive behaviors while inhibiting feeding. Thus, GnRH II can play a pivotal role in integrating nutritional status and reproduction, which is crucial for survival. This participation of the GnRH system in the relationship between nutrition and reproduction appears to have ancient evolutionary origins, as we have found that a GnRH receptor orthologue is expressed in the neurosecretory motor neuron in Caenorhabditis elegans, which integrates feeding and reproduction (Swanson et al., unpublished).
These studies suggested that GnRH II may also be effective in primates. Studies on the effects of GnRH II on proceptive female behavior in ovariectomized female marmosets109 indicated that intracerebral infusion of GnRH II at 1 microgram and 10 microgram doses stimulated proceptive behaviors in the absence of estrogen replacement. In the presence of estrogen replacement, the peptide was found to be less effective. Thus, similar to the effect of feeding in the musk shrew, estrogen may be activating the GnRH II system, and exogenous GnRH II is less effective. As in the musk shrew experiment, it was found that a GnRH antagonist, which inhibits the type I GnRH receptor but acts as an agonist at the type II GnRH marmoset receptor, stimulated proceptive behavior in the female marmoset, while GnRH I was ineffective. Thus, the pharmacology of GnRH analogue effects on reproductive behavior is distinctly different from effects on stimulating gonadotropin. For the marmoset, this difference is due to the involvement of two distinct receptors (type I and type II), which have been cloned and shown to exhibit different pharmacology. The musk shrew appears to be using a similar system, as both receptors are present (Morgan et al., unpublished). In contrast, the aberrant pharmacology for GnRH analogue effects on behavior in the rat described many years ago100 cannot be the result of using a type II GnRH receptor, as it has been deleted from the rat genome.95 It therefore appears that a different pharmacology can be manifested at the type I receptor because of the intracellular context (see ligand-induced-selective-signaling, below).
DIRECT INHIBITION OF CANCER CELL GROWTH
GnRH agonists are widely employed in the treatment of sex hormone–dependent cancers. Long-term administration of these agonists induces the desensitization of pituitary gonadotropes, resulting in a decrease in sex steroid hormone secretion and amelioration of the disease.85–91 However, many studies also implicate direct inhibitory effects of GnRH analogues in cancer cells.1,91,110
Responses of postmenopausal women with breast cancer to GnRH analogues111 suggested that GnRH analogues have direct antiproliferative effects that are independent of their actions in decreasing sex steroid hormones. The presence of GnRH receptors in breast cancer tissue112 and the demonstration of antiproliferative actions of GnRH analogues in breast cancer cell lines113–115 supported this interpretation. Antiproliferative effects of GnRH analogues and the expression of GnRH receptors have now been demonstrated in a number of cell lines of reproductive tract tumors, including prostate, uterine, and ovarian cancers, and also in non–reproductive tract tumors.110,115–119 In contrast to GnRH actions at the gonadotrope, which are mediated through the Gαq protein, these antiproliferative and apoptotic effects on tumor cells are thought to be mediated via the Gαi protein,120–122 focal adhesion complexes involving c-Src, and the JNK and P38 stress-activated kinases.118–122 Other mechanisms that have been implicated in the antiproliferative effects include the downregulation of growth factor actions by decreased expression of growth factors and their receptors and activation of phosphotyrosine phosphatase,120 and the inhibition of Akt and the 60s acidic ribosomal phosphoproteins, which modulate cell survival and protein synthesis.123–128 The effects of a series of GnRH analogues on antiproliferation and apoptosis in cancer cell lines and model HEK293 cells stably expressing the GnRH receptor are very different from their effects on gonadotropin secretion.122 This phenomenon has been described as “ligand-induced-selective signaling” (LiSS)66,122,129 (see Fig. 115-4). Thus GnRH II is more potent than GnRH I in the inhibition of cell growth, and GnRH II is less potent than GnRH I in stimulating the Gαq pathway for gonadotropin secretion.1,130–132 An extreme difference is the pure antagonism of antagonist 135-25 at Gαq while it acts as a full agonist in inhibiting cell growth.122 A number of antagonists of gonadotrope GnRH receptors act as GnRH agonists in the inhibition of tumor cell lines.113,122,133–135
Because antiproliferative and apoptotic effects are mediated by different signaling pathways from those stimulating gonadotropins, structure-activity relationship (SAR) studies have been undertaken on the inhibition of cell number by GnRH analogues using a model system of HEK 293 cells expressing the rat or the human GnRH receptor. Because GnRH II is more potent than GnRH I, the three different amino acids in GnRH II (His,5 Trp,7 and Tyr8) were systematically incorporated into synthetic GnRH I peptides, and the relative antiproliferative and inositol phosphate production (Gαq) compared. Studies revealed that the substitution of Arg8 in GnRH I by Tyr8 was the single change that resulted in the most selective inhibitor of cell growth.136 At the rat GnRH receptor, [Tyr8]GnRH I was >30-fold less potent in IP generation but 4-fold more potent in inhibiting cell growth, when compared with GnRH I.
A possible explanation for the enhanced selective inhibition of cell growth is that the substitution of Arg8 with Tyr8 enables the ligand and the receptor to adopt new conformations that better stabilize the receptor in the active state, which mediates the antiproliferative/apoptotic effect. Docking of GnRH I and GnRH II with the GnRH receptor model137 revealed that Arg8 interacts with Asp302 of the receptor (see below), but when GnRH II is docked to the receptor, Tyr8 faces away from Asp302 and appears to make contact with residues in extracellular loop 2, thus potentially stabilizing a different receptor conformation.1
GnRH RECEPTOR
GnRH RECEPTOR PRIMARY STRUCTURES
The first cloning of the mammalian GnRH receptor was accomplished by Tsutsumi et al.,138 who used RNA from the αT3 mouse gonadotrope cell line. Subsequently, a large number of GnRH receptors have been cloned from many species in all the major vertebrate classes, as well as from modern representatives of protochordate progenitors of the vertebrates.66 Although only one functional type of GnRH receptor and two endogenous ligands (GnRH I and GnRH II) exist in humans, coexistence of two or three types of GnRH ligands and receptors was identified in the majority of chordate and vertebrate species.66 However, the type II GnRH receptor has been silenced by stop codons and frameshift deletions in certain mammals, including human, chimpanzee, rat, mouse, cow, and sheep.139
The GnRH receptors are classical G protein–coupled receptors (GPCRs) with seven transmembrane domains (TMs) connected by extracellular loops (ECLs) and intracellular loops (ICLs) (see Chapter 5). They are members of the Class A rhodopsin family of GPCRs and carry the hallmarks of conserved amino acid residues of the rhodopsin superfamily (Fig. 115-5).
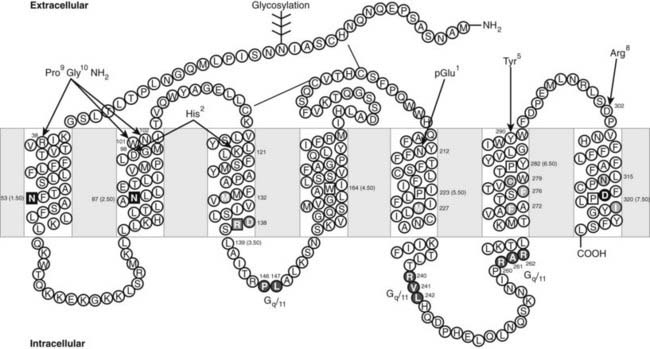
FIGURE 115-5. Two-dimensional representation of the human gonadotropin-releasing hormone (GnRH) receptor. The 7 TM domains (boxed) are connected by 3 extracellular loops (ECLs) and 3 intracellular loops (ICLs). The disulfide bonds and the glycosylation site are indicated. Residues in squares are highly conserved throughout the rhodopsin-like family of G protein–coupled receptors (GPCRs) and designated as N.50 using the nomenclature of Ballesteros and Weinstein. Ligand-binding residues are colored in red. Residues forming intramolecular interactions involved in receptor activation are grouped in different colors (green, pink, blue, and yellow). Residues involved in Gq/11 coupling are shown in purple. The intermolecular interactions between GnRH I residues and the receptor are indicated by red lines.
These conserved amino acids are used as identifiers for numbering of homologous amino acids in GPCRs of the rhodopsin family. Thus, the conserved N53 in TM1 is designated N53(1.50) and preceding amino acids 1.49, 1.48, etc., while subsequent amino acids are 1.51, 1.52, etc. Similarly the conserved N87 in TM 2 is designated N87(2.50).
Human and all mammalian pituitary GnRH receptors (designated type I GnRH receptors)66 are characterized by the unique absence among GPCRs of a carboxyterminal tail. In all other GPCRs and the GnRH receptors from mammalian (type II) and nonmammalian vertebrates and the protochordates, a carboxyterminal tail is present and interacts with intracellular proteins to elicit internalization and modulation of intracellular signaling.
Almost all GPCRs can activate G proteins, supporting the concept that GPCRs share a common protein folding and activation mechanism. The overall topology of the three-dimensional structures of rhodopsin140—the β1–141 and β2–142,143 adrenergic receptors—has been shown to be very similar. Nevertheless, structural features in the β-adrenergic receptors differ significantly from rhodopsin, which highlights the existence of receptor-specific structural features in this class of GPCRs.144 Human and other mammalian GnRH receptors have some unusual features, in addition to the absence of a carboxyterminal tail, such as a reciprocal exchange of the conserved Asp2.50-Asn7.49 pair in TM 2 and TM 7 (see Fig. 115-5). We demonstrated that mutation of Asn87(2.50) to Asp abolished receptor function, but a second mutation of Asp318(7.49) to Asn in the mouse GnRH receptor, re-creating the arrangement found in other GPCRs, restored ligand binding.145 This restoration of ligand binding by reciprocal mutation demonstrates that the side chains of the two residues in TMs 2 and 7 have complementary roles in maintaining the structure of the receptor and occupy the same microenvironment within the receptor helical bundle. Subsequent to this publication, the crystal structures of bovine rhodopsin146 and the β2-adrenergic receptor143 revealed that these residues are indeed capable of interacting through a water molecule, thus validating the GnRH receptor models. It is interesting to note that this exchange of the TM 2/TM 7 residues prevents the human GnRH receptor from coupling to phospholipase D by the small G proteins ARF and RhoA, implying receptor conformation-dependent signaling selectivity.147
GnRH RECEPTOR TERTIARY STRUCTURE
Three-dimensional structural information on the GnRH receptor has been derived from homology modeling, with the high-resolution crystal structure of bovine rhodopsin used as a template.132,137,148,149 The recently available crystal structures of β1–141, β2–142,143-adrenergic receptors and the ligand-free opsin150 have provided alternative templates for modeling the GnRH receptor. We built a homology model of the human GnRH receptor, which was refined using the experimentally identified receptor intermolecular and intramolecular interactions and energy minimization coupled with molecular dynamics simulations.132 The model reveals the experimentally identified hydrogen bonds between Asn53(1.50)-Asn87(2.50)-Asp319(7.49), between Asp98(2.61)-Lys121(3.32), and between Asp138(3.49)-Arg139(3.50) (see Figs. 115-5 and 115-6). This model has provided insights into the potential intramolecular contacts between the 7 TM domains of the human GnRH receptor, which are involved in receptor folding, tertiary structural configuration, and receptor activation. Using site-directed mutagenesis guided by molecular modeling, we have identified numerous receptor intramolecular interactions (see Fig. 115-6) that modulate receptor conformational states, accounting for ligand binding selectivity and signaling efficacy. Mutations of certain residues in the intracellular segments of TM domains to Ala (Met132(3.43), Met227(5.54), Phe272(6.40), Phe276(6.44) and Ile322(7.52)) of the human GnRH receptor allosterically increased ligand binding affinity for GnRH II but had little effect on GnRH I binding affinity.137 A similar phenotype was observed in the mutations of Cys279(6.47) and Asn315(7.45).132 We propose that these residues form part of the receptor intramolecular allosteric network (see Fig. 115-6), which transfers to specific ligand structural elements.1 Disruption of the intramolecular interactions by mutation of the network residues alters the receptor conformational state, changing ligand binding and signaling selectivity. This aspect is considered in greater depth later in the chapter.
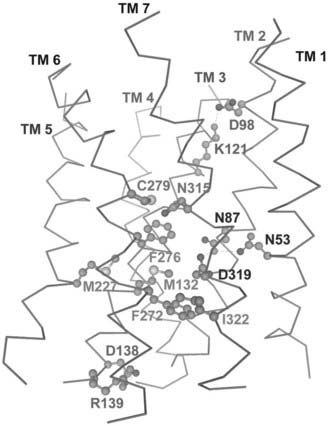
FIGURE 115-6. A model of the 7 TM domains of the human gonadotropin-releasing hormone (GnRH) receptor. The model shows experimentally identified receptor intramolecular H-bond interactions: Asn53-Asn87-Asp319, Asp98-Lys121, Asp138-Arg139, Cys279-Asn315, and the hydrophobic interactions: Met132-Phe272-Ile322 (and surrounding residues Met227 and Phe276).
LIGAND BINDING AND RECEPTOR ACTIVATION
Considerable advances in identifying putative ligand contact sites have been made by the systematic and molecular model–guided site-directed mutagenesis of amino acid residues at the mammalian GnRH receptors (see Figs. 115-5 and 115-7A).1,66,151,152 A βII′-type turn conformation of GnRH I (derived from an NMR structure; PDB code: 1YY1) was successfully docked into the receptor model, accommodating all experimentally identified receptor binding sites (see Fig. 115-7B). Molecular docking shows the intermolecular interactions between GnRH I and the receptor: pGlu1 interacts with Asn212, His2 with Lys121/Asp98, Tyr5 with Tyr290, Arg8 with Asp302, and Pro9-Gly10NH2 with Arg38/Trp101/Asn102 (see Fig. 115-7B). When GnRH II is docked into the receptor model using the same intermolecular interactions (His2 with Lys121, Pro9-Gly10NH2 with Arg38, Trp101 and Asn102, and His5 with Tyr290), Tyr8 of GnRH II (unlike Arg8 of GnRH I) faces away from Asp302 and toward ECL 2.1,136,152 We therefore propose that the different intermolecular interactions between Arg8 of GnRH I and Asp302 of ECL 3, and between Tyr8 of GnRH II and residues in ECL 2 play important roles in stabilizing different receptor active conformations with differing signaling capability (see later).
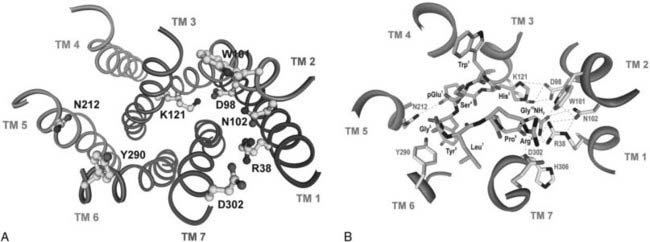
FIGURE 115-7. Gonadotropin-releasing hormone (GnRH) receptor binding sites and molecular docking of GnRH I. A, Molecular model of the human GnRH receptor showing the locations of receptor binding sites. B, A βII′ conformation of GnRH I derived from the nuclear magnetic resonance (NMR) structure (1YY1) was docked into the receptor model accommodating the experimentally identified intermolecular interactions between GnRH and the receptor. pGlu1 interacts with Asn212, His2 with Lys121/Asp98, Tyr5 with Tyr290, Arg8 with Asp302, and Pro9Gly10NH2 with Arg38/Trp101/Asn102.
Molecular mechanisms underlying ligand-mediated receptor activation have been only partially elucidated for GPCRs. A full understanding of the structural basis of GPCR activation will require high-resolution structures of active receptor conformational states with an agonist and its G protein bound.143 Briefly, initial binding of an agonist to receptor induces receptor structural rearrangement of the 7 TM helices, such as rotation and outward movements, which leads to the exposure of interacting sites of the receptor at the cytoplasmic surface to the intracellular effector proteins, including G proteins or non–G protein signaling partners. A predominant movement was observed in TM 6 with approximately 30 degrees of rotation and an 8 Å outward movement.153 The highly conserved Trp6.48 (and surrounding residues) in β2-adrenergic receptor was proposed to serve as a rotamer “toggle” switch that modulates the TM 6 Pro-kink.154 The crystal structure of the ligand-free opsin shows prominent structural changes in the conserved E/DRY of TM 3 and NPxxY of TM 7 and TM 5, 6, and 7. Consistent with previous observations,155 the intracellular end of TM 6 is tilted outward by 6 to 7 Å. Thus some of the structural changes in opsin are contributed to an active GPCR state.150
Analysis of mutations that affect GnRH receptor function has provided insight into the mechanism of receptor activation. Mutations of Met132(3.42) and Phe272(6.40) (and surrounding residues) decrease receptor expression but markedly increase mutant receptor signaling efficacy, indicating that these residues make intramolecular contacts that stabilize the receptor in the inactive state but are broken during receptor activation.137 A similar role was proposed for Cys279(6.47) and Asn315(7.45).132 The equivalent residue of Cys279(6.47) has been shown to be involved in the rotamer “toggle” switch of the β2-adrenergic receptor (see above).154 Mutation of Asp138(3.49) in the E/DRY motif of the GnRH receptor also increases receptor signaling efficacy,156 indicating an important role of this residue in receptor activation, consistent with observations of the structural change of this region in the crystal structure of opsin.150 In contrast to those that have increased mutant receptor signaling efficacy, mutations of certain residues significantly impair agonist activation (uncoupling mutations). These include Asn87(2.50), Arg139(3.50), Asp319(7.49), and Tyr323(7.53), suggesting that they form intramolecular interactions, which stabilize the receptor in the active state. Apparently, GnRH binding to receptor triggers rearrangement of the receptor intramolecular interactions, which leads to receptor activation.
RECEPTOR COUPLING
GnRH agonist occupancy of GnRH receptors leads to activation of multiple signal transduction pathways (Fig. 115-8). In gonadotropes, GnRH activates phospholipase Cβ (PLCβ) via Gq/11α, resulting in the hydrolysis of membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), which mobilize intracellular calcium and activate PKC, respectively. These in turn stimulate the biosynthesis and secretion of the gonadotropins, LH and FSH.
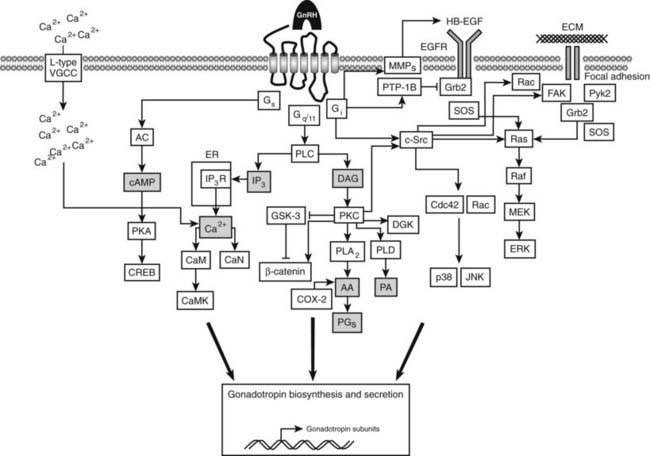
FIGURE 115-8. Gonadotropin-releasing hormone (GnRH) receptor–activated pathways. In the pituitary gonadotrope, the primary pathway involves the activation of phospholipase C (PLC) via Gq/11, resulting in the hydrolysis of membrane phosphatidylinositol 1,4-bisphosphate (PIP2) to diacylglycerol (DAG), which activates protein kinase C (PKC), and to inositol 1,4,5-trisphosphate (IP3), which triggers Ca2+ release from the endoplasmic reticulum (ER) by activating IP3 receptors (IP3R). GnRH receptor occupancy by GnRH also leads to the activation of plasma membrane L-type voltage-gated Ca2+ channels. PKC activation results in the activation of the numerous signaling cascades that lead to the activation of extracellular signal–regulated kinase (ERK), p38 mitogen–activated protein kinase (p38), and c-Jun N-terminal kinase (JNK), and to gonadotropin subunit gene transcription. The rise in intracellular Ca2+ leads to their exocytosis. The GnRH receptor can also couple to Gs, leading to the activation of adenyl cyclase (AC), a rise in cyclic adenosine monophosphate (cAMP) levels, and the activation of protein kinase A (PKA) and cAMP response element-binding protein (CREB), which also plays a role in gonadotropin subunit gene transcription. In cancer cells, coupling to Gi has been demonstrated to enhance the activities of p38 and JNK, leading to cell apoptosis. In some cell types, GnRH receptor occupancy can lead to transactivation of the EGF receptor (EGFR). Signaling via focal adhesion complexes in response to GnRH to promote cytoskeletal reorganization has been demonstrated. (See text for in-depth description.)
Gq/11 is the predominant G protein coupled to the GnRH receptor in various cellular environments.157–159 Several studies have demonstrated that other G proteins can mediate the actions of GnRH receptors. Pretreatment of rat pituitary cells with pertussis toxin decreased inositol phosphate production in response to GnRH, suggesting coupling to Gi or Go.160 In addition, GnRH receptor coupling to Gi has been demonstrated in ovarian carcinomas,120,121,161 uterine leiomyosarcomas,121 uterine endometrial carcinomas,120,162 and human prostate cancer cells.119 Pretreatment of rat pituitary cells with cholera toxin results in an increase in GnRH stimulation of LH, suggesting coupling to Gs.163,164 In addition, Gs and Gi coupling had been revealed by the GnRH stimulation of cAMP in a number of experimental paradigms.165–170
What are the structural features of the GnRH receptor that facilitate coupling to different heterotrimeric G proteins? The intracellular loops and the carboxyterminal tail have been implicated in specific coupling of GPCRs to G proteins, but their degree of involvement varies among different receptors. Because all mammalian GnRH receptors lack a carboxyterminal tail, effective receptor–G protein interactions must take place via one or more of the intracellular loops. Conservation of the carboxyterminal sequence of IC3 in vertebrate GnRH receptors suggested that this region may be crucial for coupling to the primary mediator, Gq/11. A series of cassette substitutions covering the entire sequence of IC3 confirmed this hypothesis (Wakefield, unpublished). Within this region, Ala6.29(261) was identified as an important residue for coupling. When the equivalent Ala in IC3 of biogenic amine receptors is mutated to large residues, the receptors are constitutively active.171 However, mutation of Ala6.29(261) to bulky amino acids resulted in an opposite effect in the GnRH receptor, namely, uncoupling of the receptor and failure to generate inositol phosphate.172 Mutation of the evolutionarily conserved adjacent basic amino acid (Arg6.30(262)) to Ala was also shown to result in uncoupling (Wakefield, unpublished), and natural mutations of Arg6.30(262) have been shown to cause uncoupling in receptors of families with hypogonadotropic hypogonadism.173–175 The effects of overexpression of rat GnRH receptor IC3 peptides in GnRH receptor–expressing GGH3 cells on inositol phosphate production and cAMP accumulation demonstrated that this domain is involved in coupling to Gq/11 and Gs signal transduction pathways.176
The GnRH receptors have the conserved motif DRxxxI/VxxPL at the N-terminus of IC2, which plays a role in coupling. The importance of the DRxxxI element in receptor activation appears to extend to the Pro3.57(146)Leu3.58(147). Mutation of the preceding Arg3.56(145) to Pro causes uncoupling,177 presumably because this mutation introduces a Pro-Pro motif known to disrupt secondary structure. Recently, the importance of this motif in GPCR function was demonstrated in GPR54 and in the α1A-adrenergic receptor.178 Replacement of the conserved Leu3.58(147) with Asp or Ala led to defective Gq/11 coupling,179 and mutation of Arg3.50(139) of the mouse GnRH receptor to Gln produced a similar effect.180
In IC1, the sequence (KKLSR) is a Gs recognition motif (BBxxB, where B is a basic amino acid); mutation of certain of these residues leads to uncoupling of cyclic adenosine monophosphate (cAMP) production but not of inositol phosphate production.167
Thus, the GnRH receptor is apparently able to couple to several G proteins and activate a number of effectors via different elements of the three intracellular loops. It appears that coupling to Gq/11 occurs through IC2 and IC3, and to Gs through IC1. Coupling to Gi is less clearly understood but may be related to cell type, stage of the cell cycle, or availability of the Gi protein. It has been suggested that Gi activation underlies the antiproliferative effects of GnRH in many cancers.122,161,181 The intracellular loop elements required for Gi coupling have not been investigated.
The carboxyterminal tail of GPCRs has been implicated in the regulation of signaling via receptor-coupled G proteins. In contrast to the mammalian type I GnRH receptors, but in common with other GPCRs, the cloned nonmammalian and protochordate GnRH receptors all have a carboxyterminal tail.82,182–185 The absence of a carboxyterminal tail in mammalian GnRH receptors is correlated with a lack of rapid desensitization,186 in contrast to nonmammalian and type II tailed receptors, which exhibit rapid desensitization. A number of studies187–191 involving truncation, site-directed mutagenesis, and “carboxyl-terminal tail-swapping” have established the importance of this region for coupling, desensitization, and receptor internalization (see below). Although mammalian receptors lack the carboxyterminal tail, the carboxyterminal residues of TM 7 are important for Gq/11 effector coupling.192 Because the last four residues (YFSL) of all mammalian GnRH receptors are conserved, these residues may be important for effective mammalian GnRH receptor signal transduction.
INTRACELLULAR SIGNALING
In gonadotropes, GnRH activates phospholipase Cβ (PLCβ) via Gq/11α, resulting in the hydrolysis of membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol, which mobilize intracellular calcium and activate protein kinase C (PKC), respectively. These in turn stimulate the biosynthesis and secretion of LH and FSH. Through coupling to Gq/11 and PKC activation, GnRH receptor occupancy may lead to the activation of the four key mitogen-activated protein kinase (MAPK) subfamilies, which include the extracellular signal–regulated protein kinase (ERK), the c-Jun N-terminal protein kinase (JNK), the p38 MAPK, and the big MAPK (BMK) cascades and has been the subject of numerous comprehensive reviews.193–195 In recent studies, it has been demonstrated in LβT2 cells that ERK activation induces c-fos and LHβ gene expression, and that this signaling process occurs by GnRH receptor activation of both Gq/11 and Gs,169,196,197 further implicating coupling of the GnRH receptor to multiple G proteins and the activation of different MAPK signaling cascades to regulate selective gonadotropin subunit expression. Activation of ERK in LβT2 cells may regulate basal and GnRH-induced α subunit and LHβ subunit expression, and JNK activation regulates basal and GnRH-stimulated LHβ subunit expression—both ERK and JNK involving c-Src participation (see reference 196 and references therein). The rise in intracellular calcium levels in response to GnRH is important in the regulation of LHβ subunit expression. This was demonstrated by a study in LβT2 cells showing that GnRH induces the activation of calcium/calmodulin-dependent kinase type II (Ca/CaMK II), which is mediated by the elevation of intracellular calcium levels.198 Ca/CaMK II probably acts at one or more GnRH-sensitive transcription factors at the LHβ subunit promoter to regulate LH expression. Differential regulation of LH and FSH secretion in the tilapia, a bony fish, is achieved through divergent signaling via the GnRH receptor, with expression of α subunit and LHβ subunit mRNAs being increased by activation of the ERK cascade, while induction of FSHβ transcript expression is ERK independent and is regulated by the cAMP-PKA signaling cascades.199
Although it has been demonstrated that two activator protein-1 (AP-1) elements located in the proximal FSHβ promoter are not involved in GnRH regulation of FSHβ gene expression, GnRH activation of the ERK cascade in LβT2 cells is reported to regulate FSHβ gene expression.200 GnRH activation of MAPK cascades regulates not only gonadotropin subunit gene expression, but also regulation of the GnRH receptor gene. GnRH activation of the JNK pathway regulates the activation of AP-1, which acts via the AP-1 element in the GnRH receptor promoter to drive endogenous GnRH receptor gene expression in αT3-1 cells.201
The rise in intracellular calcium levels, derived in part from IP3-activated intracellular storage pools and in part from the influx of extracellular calcium through L-type voltage-sensitive calcium channels, is of paramount importance for initiating the exocytotic events that lead to the release of gonadotropins from the gonadotrope in the anterior pituitary.159,202
The roles of both heterotrimeric and monomeric G proteins in divergent GnRH receptor signaling have recently been described.203 GnRH receptor occupancy leads to dramatic changes in cell adhesion and superstructural morphology, in the form of actin cytoskeleton remodeling, and these effects are mediated by FAK/c-Src/ERK activation at focal adhesion complexes, and are not dependent on PKC and intracellular calcium mobilization.203 In relation to this, the importance of the focal adhesion complex, and in particular the focal adhesion kinase Pyk2, in signaling via the ERK pathway to LHβ gene transcription in the gonadotrope (LβT2), has now been established.204
c-Src plays a central role in many GnRH signaling pathways. GnRH mediates a functional interaction between c-Src and the lipid kinase diacylglycerol kinase (DGK)-ζ in both HEK 293 and LβT2 cells.205 In addition, it has been demonstrated that the activation of DGK-ζ exerts a functional role in the LβT2 gonadotrope cell line, in which elevated expression of DGK-ζ resulted in shortening of the timescale of ERK activation, suggesting a potential role of endogenous DGK-ζ in controlling the induction of gonadotropin subunit gene expression, which is stimulated by GnRH via activation of ERK1/2. This mechanism may be mediated by the depletion of DAG due to its hydrolysis by DGK-ζ to phosphatidic acid after translocation to the plasma membrane.205
Arachidonic acid (AA) is converted to prostanoids (PGs) via activated cyclooxygenase (COX)-2 and specific PG synthases.206 GnRH activation of COX-2 results in PG synthesis. It has been shown that GnRH stimulates AA release from rat pituitary cells, followed by elevated expression of 12-lipoxygenase and formation of 5-lipoxygenase products, which participate in GnRH actions.207,208 A novel GnRH signaling pathway is mediated by PGF2α and the FP receptor and PGI2 and the IP receptor, which limit the homologous regulation of GnRH receptor; however, PGF2α also exerts selective inhibition of LH release.209 This mechanism may underlie the cyclic responsiveness of pituitary gonadotropes to GnRH and the asynchronous LH and FSH release during the mammalian ovarian cycle.
GnRH stimulation of the LβT2 gonadotrope cell line results in an elevated mRNA level of the BMP antagonist DAN, as well as mature cytosolic and secreted DAN glycoprotein.210 Overexpression of DAN in LβT2 cells inhibited the synergistic activation of GnRH receptor gene expression by GnRH and activin, but had no effect on GnRH- or activin-stimulated gonadotropin gene expression, thus revealing an additional signaling mechanism for the coordinated regulation of gonadotropin biosynthesis and secretion; this is crucial for the asynchronous secretion of LH and FSH during the ovarian cycle.
The ability of GnRH analogues to inhibit proliferation of androgen-dependent tumor cells suggests that GnRH is able to modulate androgen receptor (AR) activity. In fact, a GnRH-induced complex of the focal adhesion kinase Pyk2, with c-Src and the AR cofactor Hic-5, has been demonstrated to directly modulate AR subcellular location and nuclear activity, such that the AR is rendered transcriptionally inactive in response to GnRH.211 This scaffold-dependent signaling of the GnRH receptor to the AR is potentially involved in the direct antiproliferative effects of GnRH analogues on prostatic neoplasms and cell lines.
A role for β-catenin as a member of a transcription factor complex that drives maximal activity of the LHβ promoter in response to GnRH has been demonstrated.212 The demonstrated co-localization of β-catenin with SF1 and EGR1 on the promoter of the LHβ subunit gene in response to GnRH suggested that endogenous SF-1 and β-catenin can physically associate in LβT2 cells. Stabilization and nuclear accumulation of β-catenin is a preeminent hallmark of the Wnt/β-catenin signaling pathway. The seminal demonstration of GPCR activation of β-catenin/TCF-mediated transcriptional activity, which was reported for the prostanoid FPB receptor, suggested that this may be a property of other GPCRs.213,214 In fact, β-catenin accumulation in the nucleus, activation of a TCF-Luciferase reporter, and upregulation of Wnt target genes in response to GnRH in gonadotrope cells have been demonstrated.215 Thus, β-catenin signaling in response to GnRH stimulation in LβT2 gonadotrope cells is important not only as a cofactor for TCF/LEF-dependent transcriptional activity at Wnt target genes, but also in its important role in mediating gonadotropin gene expression.212 Furthermore, results from the study by Gardner et al. demonstrate that GnRH stimulates β-catenin/TCF signaling in a heterologous cell system, which has implications for GnRH influencing Wnt/β-catenin signaling processes in a variety of peripheral tissues and cancers that express the type I GnRH receptor. The activation of Wnt target genes by GnRH demonstrates the ability of GnRH to influence the output of Wnt signaling and poses the question, “Why does GnRH activate the same target genes as Wnt ligands?”
In addition to heterotrimeric and monomeric G proteins, the involvement in GnRH receptor–mediated signaling of regulators of G protein signaling (RGS) proteins has been shown. RGS proteins interact directly with active Gα subunits to accelerate their intrinsic GTPase activity and limit their half-life.216 RGS3 and RGS10 have been implicated in the regulation of GnRH receptor coupling,217,218 and evidence suggests that the carboxyterminal tail of nonmammalian GnRH receptors may contain elements for interactions with RGS10, although the nature of this interaction is unclear.218
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


