FIGURE 100-1. The structures of commonly used glucocorticoids. In the depiction of cortisol, the 21 carbon atoms of the glucocorticoid skeleton are indicated by numbers, and the four rings are designated by letters. The arrowheads indicate the structural differences between cortisol and each of the other molecules.
(Data from Axelrod L: Glucocorticoid therapy. Medicine [Baltimore] 55:39–65, 1976.)
Pharmacodynamics
HALF-LIFE, POTENCY, AND DURATION OF ACTION
The important differences among the systemically used glucocorticoid compounds are duration of action, relative glucocorticoid potency, and relative mineralocorticoid potency1,2 (Table 100-1). The commonly used glucocorticoids are classified as short-acting, intermediate-acting, and long-acting on the basis of the duration of adrenocorticotropic hormone (ACTH) suppression after a single dose, equivalent in antiinflammatory activity to 50 mg of prednisone5 (see Table 100-1). The relative potencies of the glucocorticoids correlate with their affinities for the glucocorticoid receptor.6 The observed potency of a glucocorticoid, however, is determined not only by the intrinsic biological potency but also by the duration of action.6,7 The relative potency of two glucocorticoids varies as a function of the time interval between the administration of the two steroids and the determination of the potency. In particular, failure to consider the duration of action may lead to a marked underestimation of the potency of dexamethasone.7
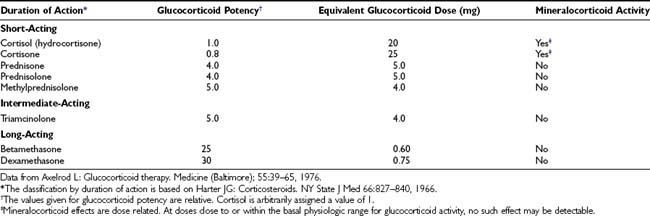
Little correlation exists between the circulating half-life (T1/2) of a glucocorticoid and its potency. The T1/2 of cortisol in the circulation is 80 to 115 minutes.1 The T1/2 values of other commonly used agents are as follows: cortisone, 0.5 hour; prednisone, 3.4 to 3.8 hours; prednisolone, 2.1 to 3.5 hours; methylprednisolone, 1.3 to 3.1 hours; and dexamethasone, 1.8 to 4.7 hours.1,7,8 Although prednisolone and dexamethasone have comparable T1/2 values, dexamethasone is clearly more potent. Similarly, little correlation is found between the T1/2 of a glucocorticoid and its duration of action. The many actions of glucocorticoids do not have an equal duration.
The duration of action is a function of the dose. For example, the duration of ACTH suppression produced by an individual glucocorticoid is dose related.5 The duration of ACTH suppression is not simply a function of the level of antiinflammatory activity, because variations in the duration of ACTH suppression are achieved by doses of glucocorticoids with comparable antiinflammatory activity.
In short, the slight differences in the T1/2 values of the glucocorticoids contrast with their marked differences in potency and duration of ACTH suppression. Thus the potency and the duration of action of a glucocorticoid are not determined solely by its presence in the circulation; this is consistent with the mechanism of action of steroid hormones. Steroid molecules bind to a specific intracellular receptor protein, the glucocorticoid receptor. The steroid-receptor complex modifies the process of transcription by which RNA is transcribed from the DNA template, among other mechanisms of action (see later discussion). This process alters the rate of synthesis of specific proteins. The steroid thereby modifies the phenotypic expression of the genetic information. Thus a glucocorticoid continues to act inside the cell after it has disappeared from the circulation. Moreover, the events initiated by a glucocorticoid may continue to occur, or a product of these events (such as a specific protein) may be present, after the disappearance of the glucocorticoid from the circulation.
BIOAVAILABILITY, ABSORPTION, AND BIOTRANSFORMATION
Normally the plasma cortisol level is much lower after oral administration of cortisone than after an equal dose of cortisol.9 Although oral cortisone may be adequate replacement therapy in chronic adrenal insufficiency, the oral form of this agent should not be used when pharmacologic effects are sought. Comparable plasma prednisolone levels are achieved in normal persons after equivalent oral doses of prednisone and prednisolone.8,10 After the administration of either of these substances, a wide variation is found in individual prednisolone concentrations, which may reflect variability in absorption.8
In contrast to the marked increase in the plasma cortisol level that follows the intramuscular injection of hydrocortisone, the plasma cortisol level increases little or not at all after an intramuscular injection of cortisone acetate. When given intramuscularly, cortisone acetate does not provide an adequate plasma cortisol level and offers no advantage over hydrocortisone delivered by the same route. The explanation for the failure of intramuscular cortisone acetate to provide adequate plasma cortisol levels is not known. It may reflect poor absorption from the site of injection. Intramuscular cortisone acetate, which reaches the liver through the systemic circulation, also may be metabolically inactivated before it can be converted to cortisol in the liver, in contrast to oral cortisone acetate, which reaches the liver through the portal circulation.
PLASMA TRANSPORT PROTEINS
In normal people, circadian fluctuations occur in the capacity of corticosteroid-binding globulin (transcortin) to bind cortisol and prednisolone. Patients who have received prednisone for a prolonged period have no diurnal variation in the binding capacity of corticosteroid-binding globulin for cortisol or prednisolone, and both capacities are reduced in comparison with normal persons. Thus, long-term glucocorticoid therapy not only changes the endogenous secretion of steroids but also affects the transport of some glucocorticoids in the circulation. This may explain why the disappearance of prednisolone from the circulation is more rapid in those persons who have previously received glucocorticoids.
Glucocorticoid Therapy in the Presence of Liver Disease
Plasma cortisol levels are normal in patients with liver disease. Although the clearance of cortisol is reduced in patients with cirrhosis, the hypothalamopituitary-adrenal (HPA) homeostatic mechanism remains intact. Consequently, the decreased clearance rate is accompanied by decreased synthesis of cortisol.
The conversion of prednisone to prednisolone is impaired in patients with active liver disease.11 This is offset in large part by a decreased rate of elimination of prednisolone from the plasma.11 In patients with liver disease, the plasma availability of prednisolone is quite variable after oral doses of either prednisone or prednisolone.11 The percentage of plasma prednisolone that is bound to protein is reduced in patients with active liver disease; the unbound fraction is inversely related to the serum albumin concentration. The frequency of prednisone side effects is increased at low serum albumin levels.12 Both findings may reflect impaired hepatic function. Because the impairment of conversion of prednisone to prednisolone is quantitatively small in patients with liver disease and is offset by the decreased clearance rate of prednisolone, and because of the marked variability in plasma prednisolone levels after the administration of either corticosteroid, no clear mandate exists to use prednisolone rather than prednisone in patients with active liver disease or cirrhosis.8 If prednisone or prednisolone is used, however, a dose somewhat lower than usual should be given if the serum albumin level is low.8
Glucocorticoid Therapy and the Nephrotic Syndrome
When hypoalbuminemia is caused by the nephrotic syndrome, the fraction of prednisolone that is protein bound is decreased. The unbound fraction is inversely related to the serum albumin concentration. The unbound prednisolone concentration remains normal, however.13,14 Because the pharmacologic effect is determined by the unbound concentration, altered prednisolone kinetics do not explain the increased frequency of prednisolone-related side effects in patients with the nephrotic syndrome.
Glucocorticoid Therapy and Hyperthyroidism
The bioavailability of prednisolone after an oral dose of prednisone is reduced in patients with hyperthyroidism because of decreased absorption of prednisone and increased hepatic clearance of prednisolone.15
Glucocorticoids During Pregnancy and in the Early Postnatal Period
Glucocorticoid therapy is well tolerated by the mother in pregnancy.16 Glucocorticoids cross the placenta, but no compelling evidence indicates that this produces clinically significant adrenal insufficiency or Cushing’s syndrome in the neonate, although subnormal responsiveness to exogenous ACTH may occur.16 Maternal glucocorticoid exposure during the first 8 weeks after conception is associated with an increased risk of cleft lip and palate but not cleft palate alone.17 Otherwise, no evidence exists that glucocorticoids during pregnancy increase the incidence of congenital defects in humans.16 Studies in animals indicate that maternal treatment with glucocorticoids programs the offspring, causing increases in blood pressure, glucose levels, HPA activity, and anxiety-related behaviors.18 In humans, antenatal glucocorticoid therapy may be associated with hypertension in adolescence, hyperinsulinemia, and subtle effects on neurologic function in offspring.18 Glucocorticoids during pregnancy decrease the birth weight of term infants; the long-term consequences of this are unknown. Glucocorticoid therapy initiated within the first 12 hours after birth is followed by reduced stature and head circumference and impaired neuromotor and cognitive function at school age.19,20 Because the concentrations of prednisone and prednisolone in breast milk are low, the administration of these drugs to the mother of a nursing infant is unlikely to produce deleterious effects in the infant.
Glucocorticoid Therapy and Aging
The clearance of prednisolone and methylprednisolone decreases with age.21,22 Although prednisolone levels are higher in elderly subjects than in young subjects after comparable doses, endogenous plasma cortisol levels are suppressed to a lesser extent in the elderly.21 These findings may be associated with an increased incidence of side effects and suggest the need to use smaller doses in the elderly than in young patients.
Drug Interactions
The concomitant use of other medications can alter the effectiveness of glucocorticoids; the reverse also is true.23
EFFECTS OF OTHER MEDICATIONS ON GLUCOCORTICOIDS
The metabolism of glucocorticoids is accelerated by substances that induce hepatic microsomal enzyme activity, such as phenytoin, barbiturates, and rifampicin. The administration of these medications can increase the corticosteroid requirements of patients with adrenal insufficiency or lead to deterioration in the condition of patients whose underlying disorders are well controlled by glucocorticoid therapy. These substances should be avoided if possible in patients receiving corticosteroids. Diazepam does not alter the metabolism of glucocorticoids and is preferable to barbiturates in this setting. If drugs that induce hepatic microsomal enzyme activity must be used in patients taking corticosteroids, an increase in the required dose of corticosteroids should be anticipated.
Conversely, ketoconazole increases the bioavailability of large doses of prednisolone (0.8 mg/kg) because of inhibition of hepatic microsomal enzyme activity.24 Oral contraceptive use decreases the clearance of prednisone and increases its bioavailability.25
The bioavailability of prednisone is decreased by antacids in doses comparable to those used clinically.26 The bioavailability of prednisolone is not impaired by sucralfate, H2-receptor blockade, or cholestyramine.
EFFECTS OF GLUCOCORTICOIDS ON OTHER MEDICATIONS
The concurrent administration of a glucocorticoid and a salicylate may reduce the serum salicylate level. Conversely, reduction of the glucocorticoid dose during the administration of a fixed dose of salicylate may produce a higher and possibly toxic serum salicylate level. This interaction may reflect the induction of salicylate metabolism by glucocorticoids.27
Glucocorticoids may increase the required dose of insulin or other agents used to treat hyperglycemia, blood-pressure medications, or glaucoma medications. They also may alter the required dose of sedative-hypnotic or antidepressant therapy. Digitalis toxicity can result from hypokalemia caused by glucocorticoids, as from hypokalemia of any cause. Glucocorticoids can reverse the neuromuscular blockade induced by pancuronium.
Considerations Before Initiating Use of Glucocorticoids as Pharmacologic Agents
Cushing’s syndrome is a life-threatening disorder. The 5-year mortality rate was more than 50% at the beginning of the era of glucocorticoid and ACTH therapy.28 Infections and cardiovascular complications were frequent causes of death. High-dose exogenous glucocorticoid therapy is similarly hazardous.
Table 100-2 presents the important questions to consider before initiating glucocorticoid therapy.29 These questions enable the physician to assess the potential risks of treatment that must be weighed against the possible benefits. The more severe the underlying disorder, the more readily systemic glucocorticoid therapy can be justified. Thus, corticosteroids are commonly used in patients with severe forms of systemic lupus erythematosus, sarcoidosis, active vasculitis, asthma, transplantation rejection, pemphigus, or diseases of comparable severity. Systemic corticosteroids should not be administered to patients with mild bronchial asthma, who should receive more conservative therapy first, including inhaled glucocorticoids.30 Inhaled glucocorticoids are the most effective long-term control medication for asthma across all age groups.30
Table 100-2. Considerations Before the Use of Glucocorticoids as Pharmacologic Agents
6. Have other modes of therapy been used to minimize the glucocorticoid dose and to minimize the side effects of glucocorticoid therapy? |
Data from Thorn GW: Clinical considerations in the use of corticosteroids. N Engl J Med 274:775–781, 1966.
DURATION OF THERAPY
The anticipated duration of glucocorticoid therapy is a critical consideration. The use of glucocorticoids for 1 to 3 weeks for a condition such as poison ivy or allergic rhinitis is unlikely to be associated with serious side effects in the absence of a contraindication. An exception to this rule is a corticosteroid-induced psychosis, which may occur after only a few days of high-dose glucocorticoid therapy, even in patients with no previous history of psychiatric disease.31,32 The risk of most complications is related to the dose and duration of therapy.33–36 Consequently, one should prescribe the smallest possible dose for the shortest possible period. If hypoalbuminemia is present, the dose should be reduced. If long-term treatment is indicated, the use of alternate-day glucocorticoid therapy should be considered (see later).
LOCAL USE
Local corticosteroid preparations should be used whenever possible because they produce fewer side effects than do systemically administered agents. Examples include topical therapy in dermatologic disorders such as bullous pemphigoid, corticosteroid aerosols in bronchial asthma and allergic rhinitis, and corticosteroid enemas in ulcerative proctitis.30,37,38 Inhaled glucocorticoids have a markedly better safety profile than do orally administered agents.39 Nevertheless, adrenal suppression and other complications may develop from topical, inhaled, and regional administration of glucocorticoids. The risk factors for the development of these side effects from topical steroids for dermatologic indications include application to a large surface area of skin, application for a prolonged period, use of occlusive dressings, and use of a highly potent (class I) glucocorticoid. In sufficient doses, inhaled glucocorticoids produce an acute but apparently temporary decrease in growth velocity in children, cause osteoporosis, and may increase the risk of cataracts, glaucoma, skin atrophy, and ecchymoses.39 The development of adrenal suppression from inhaled steroids is related to dose, duration of therapy, and use of a potent agent (e.g., fluticasone).40 The intraarticular injection of corticosteroids may be of value in carefully selected patients if strict aseptic techniques are used and if frequent injections are avoided.
SELECTING A SYSTEMIC PREPARATION
Agents with no mineralocorticoid activity should be used when a glucocorticoid is prescribed for pharmacologic purposes. If the dosage is to be tapered over a few days, a long-acting agent should be avoided. For alternate-day therapy, one should use a short-acting agent that generally does not cause sodium retention (e.g., prednisone, prednisolone, or methylprednisolone). The use of supplemental medications to minimize the systemic corticosteroid dose and to reduce the side effects of systemic glucocorticoids should always be considered. In asthma, for example, treatment should include inhaled glucocorticoids and bronchodilators such as β-adrenergic agonists and theophylline.
Effects of Exogenous Glucocorticoids
ANTIINFLAMMATORY AND IMMUNOSUPPRESSIVE EFFECTS
Among their many actions, endogenous glucocorticoids protect the organism from damage caused by its own defense reactions and the products of these reactions during stress. They do this by confining inflammatory responses to the area of injury. Consequently, the use of glucocorticoids as antiinflammatory and immunosuppressive agents represents an application of the physiologic effects of glucocorticoids to the treatment of disease (see Chapter 98).
Chronic inflammation is characterized by the increased expression of multiple inflammatory genes that are regulated by transcription factors such as nuclear factor (NF)-κB and activator protein-1 (AP-1).41–45 These transcription factors bind to and activate coactivator molecules, which acetylate core histone proteins, causing the DNA to unwind, thereby switching on gene transcription, a process called chromatin remodeling.44,45 Glucocorticoids switch off multiple inflammatory genes (including those related to synthesis of cytokines, chemokines, adhesion molecules, inflammatory enzymes, receptors, and other proteins) that are activated during the chronic inflammatory process.41–45 They do so principally by reversing histone acetylation of activated inflammatory genes through binding of glucocorticoid receptors to coactivators (which have been activated by transcription factors such as NF-κB and AP-1) and recruitment of histone deacetylase-2 to the activated transcription site.44,45 In high concentrations, glucocorticoids increase the synthesis of antiinflammatory proteins, notably annexin-1 (also called lipocortin-1), an inhibitor of phospholipase A2 (see following); inhibit transcription of genes linked to proteins related to the pathogenesis of glucocorticoid side effects; and have postgenomic effects.44,45
Influence on Blood Cells and on Microvasculature
Glucocorticoid effects on inflammatory and immune phenomena include effects on leukocyte movement, leukocyte function, and humoral factors. In general, glucocorticoids have a greater effect on leukocyte traffic than on function and more effect on cellular than on humoral processes.46,47 Glucocorticoids alter the traffic of all the major leukocyte populations in the circulation.
Perhaps the most important antiinflammatory effect of glucocorticoids is the ability to inhibit the recruitment of neutrophils and monocyte-macrophages to an inflammatory site.47 Glucocorticoids modify the increased capillary and membrane permeability that occurs in an area of inflammation. By decreasing the microvasculature dilatation and increased capillary permeability that occur during an inflammatory response, the exudation of fluid and the formation of edema may be reduced, and the migration of leukocytes may be impaired.2,47,48 The decrease in the accumulation of inflammatory cells also is related to decreased adherence of inflammatory cells to the vascular endothelium. This may reflect decreased expression of adhesion molecules E-selectin and intercellular adhesion molecule 1 (ICAM-1) by stimulated endothelial cells.49
Glucocorticoids have many effects on leukocyte function.47 Glucocorticoids suppress cutaneous delayed hypersensitivity responses. Monocyte-macrophage traffic and function are sensitive to glucocorticoids. Glucocorticoids, in divided daily doses, depress the bactericidal activity of monocytes. The sensitivity of monocytes to glucocorticoids may explain the effectiveness of these agents in many granulomatous diseases, because the monocyte is the principal cell involved in granuloma formation.47 Although neutrophil traffic is sensitive to glucocorticoids, neutrophil function appears to be relatively resistant to these agents.47 Whereas most in vivo studies of neutrophil phagocytosis have found no evidence for impairment of phagocytosis or bacterial killing,47 other studies suggest that glucocorticoids induce a generalized phagocytic defect, affecting both granulocytes and monocytes.
Glucocorticoid therapy retards the disappearance of sensitized erythrocytes, platelets, and artificial particles from the circulation.47 This may account for the efficacy of glucocorticoids in the treatment of idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia.
Influence on Arachidonic Acid Derivatives
Glucocorticoids inhibit prostaglandin (PG) and leukotriene synthesis by inhibiting the release of arachidonic acid from phospholipids.50 The inhibition of arachidonic acid release appears to be mediated by the induction of annexin-1 (lipocortin-1) and other lipocortins, a family of related proteins that inhibit phospholipase A2, an enzyme that liberates arachidonic acid from phospholipids.51,52 This mechanism is distinct from the mechanism of action of the nonsteroidal antiinflammatory agents, such as salicylates and indomethacin, which inhibit the cyclooxygenase that converts arachidonic acid to the cyclic endoperoxide intermediates in the PG synthetic pathway; in some tissues, glucocorticoids inhibit cyclooxygenase activity. Thus the glucocorticoids and the nonsteroidal antiinflammatory agents exert their antiinflammatory effects at two distinct but adjacent loci in the pathway of arachidonic acid metabolism. Glucocorticoids and nonsteroidal antiinflammatory agents have different therapeutic effects. Some of the therapeutic effects of glucocorticoids that are not produced by the nonsteroidal antiinflammatory agents may be related to the inhibition of leukotriene formation.50
SIDE EFFECTS
The side effects of glucocorticoids include the diverse manifestations of Cushing’s syndrome and HPA suppression36,53 (Table 100-3) Exogenous Cushing’s syndrome differs from endogenous Cushing’s syndrome in several respects. Hypertension, acne, menstrual disturbances, male erectile dysfunction, hirsutism or virilism, striae, purpura, and plethora are more common in endogenous Cushing’s syndrome. Benign intracranial hypertension, glaucoma, posterior subcapsular cataract, pancreatitis, and avascular necrosis of bone are virtually unique to exogenous Cushing’s syndrome. Obesity, psychiatric symptoms, and poor wound healing have nearly equal frequency in endogenous and exogenous Cushing’s syndrome.53,54 These differences may be explained as follows. When Cushing’s syndrome is caused by exogenous glucocorticoids, ACTH secretion is suppressed; in spontaneous, ACTH-dependent Cushing’s syndrome, the elevated ACTH output causes bilateral adrenal hyperplasia. In the former circumstance, the secretion of adrenocortical androgens and mineralocorticoids is not increased. Conversely, when ACTH output is elevated, the secretion of adrenal androgens and mineralocorticoids may be increased.1 The augmented secretion of adrenal androgens may account for the higher prevalence of virilism, acne, and menstrual irregularity in the endogenous form of Cushing’s syndrome, and the enhanced production of mineralocorticoids may explain the higher prevalence of hypertension.1 Some of the complications that are virtually unique to exogenous Cushing’s syndrome arise after the prolonged use of large doses of glucocorticoids. Examples are benign intracranial hypertension, posterior subcapsular cataract, and avascular necrosis of bone.1
Table 100-3. Adverse Reactions to Glucocorticoids
Glucocorticoids appear to increase the risk of peptic ulcer disease and also gastrointestinal hemorrhage.55 The magnitude of the association between glucocorticoid therapy and these complications is small and is related to the total dose and duration of therapy.55,56 The risk of peptic ulcer disease and related gastrointestinal problems is increased by the concurrent use of glucocorticoids and nonsteroidal antiinflammatory drugs.57,58
Glucocorticoid therapy, especially daily therapy, may suppress the immune response to skin tests for tuberculosis. When possible, a tuberculin skin test should be performed before the initiation of glucocorticoid therapy, with the intention to treat with isoniazid if the skin test meets the criteria of the American Thoracic Society.59
Some patients respond to (and experience side effects of) glucocorticoids more readily than others at comparable doses. Increased responsiveness to glucocorticoids may be a consequence of hypoalbuminemia, the nephrotic syndrome, impaired renal function, age, drug interactions, and variations in the severity of the underlying disease (see earlier). Impaired renal function may contribute to a decrease in the clearance of prednisolone and an increase in the prevalence of cushingoid features.60 In patients who experience side effects, the metabolic clearance rate of prednisolone and the volume of distribution are lower10,61 and the T1/2 is longer than in those who do not.61 Patients who have a cushingoid habitus while taking prednisolone have higher endogenous plasma cortisol levels than do those without this complication, perhaps because of resistance of the HPA axis to suppression by exogenous glucocorticoids.62 Alterations in bioavailability probably do not account for variations in the therapeutic response to glucocorticoids.
Variations in the effectiveness of corticosteroids may be the result of altered cellular responsiveness to the drugs.63–66 In patients with primary open-angle glaucoma, exogenous glucocorticoids produce a more pronounced increase of intraocular pressure63; a greater suppression of the 8:00 am plasma cortisol level when dexamethasone, 0.25 mg, is administered the previous evening at 11:00 pm;65 and greater suppression of phytohemagglutinin-induced lymphocyte transformation64,66 than in normal persons. Primary open-angle glaucoma is relatively common. These findings suggest that a distinct subpopulation of patients is hyperresponsive to glucocorticoids and that this sensitivity is genetically determined.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


