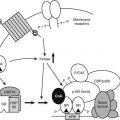
FIGURE 97-1. Outline of regulation and actions of glucocorticoids. See text for details.
Normal glucocorticoid levels are regulated in a range and time course that reflect varying physiologic needs as well as the vulnerability of the organism, particularly the brain,59 to harm from excessive exposure. Basal hormone levels follow a circadian rhythm and reach peak values before the period of daily activity.60 Their actions maintain or permissively “prime” homeostatic mechanisms and protect against moderate stress. Stress-induced levels, which can far exceed peak basal levels, appear necessary to cope with severe stress. Peak basal levels cause Cushing’s syndrome if maintained indefinitely, so circadian lowering of glucocorticoid concentrations is physiologically necessary.
Synthesis and secretion of glucocorticoids is controlled by neural and humoral signals that change throughout the day and respond to stress and negative feedback.60 The main components of this system (see Fig. 97-1) are the adrenal cortex, where glucocorticoid secretion is stimulated by ACTH; the anterior pituitary, where ACTH secretion is stimulated by CRH, vasopressin (VP), and other secretagogues, and are inhibited by glucocorticoids; and the central nervous system, where CRH and VP synthesis in the hypothalamus is stimulated by stress and other influences, and is inhibited by glucocorticoids. Paradoxically, chronic actions of glucocorticoids on the brain exerted over days can be excitatory.61
Glucocorticoids exert feedback control on pituitary corticotrophs, the paraventricular nucleus (PVN) of the hypothalamus, and probably the hippocampus.42 Synthetic analogues like dexamethasone and prednisolone are exported from the brain by a multidrug resistance efflux transporter P-glycoprotein in the blood-brain barrier, which acts predominantly on the pituitary.42,54 Glucocorticoid regulation appears to be mediated both by GRs, which are found throughout the brain with high concentrations in the PVN, and by MRs, which are located mainly in the hippocampus and lateral septum. Actions on the brain via MRs can be considered to permissively control sensitivity to rapid CRH responses via CRH-1 receptors, maintaining the capacity of the HPA axis to respond to stress and maintain homeostasis; actions through GRs restrain stress-induced responses and facilitate learning and recovery of homeostasis.58
Studies on transgenic mice with altered GRs extend these conclusions.62,63 Mice with low levels of GRs have increased levels of CRH (not VP), ACTH, and corticosterone, as well as hypertrophy of the adrenal cortex. Overexpressed GRs reverse this picture. Mice with GRs that cannot dimerize have normal CRH and ACTH, showing that feedback via GRs probably is provided through genes controlled by protein-protein cross-talk. However, the gene for pro-opiomelanocortin (POMC), the ACTH precursor, is upregulated, implying control by GR dimers, which probably bind to nGREs, as noted below.
Inactivation of GRs in the nervous system64 leads to higher levels of corticosterone with Cushing’s-like symptoms. CRH is elevated, as is ACTH in pituitary corticotrophs, but circulating ACTH levels are slightly reduced—a divergence between ACTH and corticosterone reminiscent of that seen in clinically depressed patients.
Fig. 97-1 also illustrates one side of the reciprocal relation between the immune and neuroendocrine systems.65 Cytokines like interleukin 1 (IL-1), IL-6, and tumor necrosis factor (TNF)-α, which are produced mainly in the immune system but also by brain cells, stimulate the HPA axis. IL-1, IL-6, and TNF-α are proinflammatory cytokines, so stimulation of glucocorticoid secretion limits their activity throughout the organism. The importance of IL-1, for example, is revealed in knockout mice lacking IL-1 receptors and in mice overexpressing IL-1 antagonist in the brain66: they have reduced stress responses and fail to hypersecrete ACTH after adrenalectomy. Another regulator of the HPA axis may be leptin.67 A remarkable observation is that sucrose ingestion, like glucocorticoid replacement, can restore to normal most of the consequences of adrenalectomy on feeding and metabolism, and on the HPA axis, including ACTH levels, which presumably mimic signals from the metabolic effects of glucocorticoids.68
Each stage of the HPA feedback loop will now be considered.
ADRENAL CORTEX: GLUCOCORTICOIDS
Synthesis of glucocorticoids, generally ascribed solely to the adrenal cortex, has been reported to occur in the thymus.69 It also occurs in the intestinal mucosa, where it influences local immune responses.70 In the adrenal cortex, glucocorticoid synthesis is closely tied to plasma levels of ACTH, which exhibit episodic peaks and circadian rhythm similar to plasma levels of glucocorticoids. ACTH stimulates steroidogenesis by binding to membrane receptors on adrenal cells, which activates adenylate cyclase and also causes hypertrophy and hyperplasia of the adrenal cortex. Leptin inhibits ACTH stimulation of cortisol secretion by adrenal cells.67
PITUITARY: ACTH
The synthesis and secretion of ACTH in anterior pituitary corticotrophs are stimulated by CRH and VP, modulated by catecholamines, and inhibited by glucocorticoids. CRH binds to receptors on pituitary cell membranes and activates adenylate cyclase; cAMP then stimulates both secretion and synthesis of ACTH. Activity of CRH is strongly potentiated by VP. Whereas CRH increases the amount of ACTH secreted from each responsive corticotroph, VP, probably through the phosphoinositide pathway, increases the number of CRH-responsive corticotrophs. Nonetheless, knockout mice defective in type 1 CRH receptor (CRH-R1) respond to inflammatory stress with pronounced increases in ACTH and corticosterone that do not depend critically on CRH or VP.71
Glucocorticoids inhibit ACTH secretion directly by suppressing POMC expression in pituitary corticotrophs, and indirectly by inhibiting secretion of CRH and VP.60 After adrenalectomy, ACTH secretion rises, retaining its circadian rhythm. CRH and VP levels in the PVN also rise. These and other changes are reversed by glucocorticoids. Annexin 1 (lipocortin 1), a glucocorticoid-induced protein, mediates glucocorticoid inhibition of secretion of ACTH from the pituitary, apparently through a nongenomic mechanism.
Feedback has been classified according to how rapidly it inhibits ACTH secretion60: fast (within 30 minutes of hormone administration), delayed (minutes to hours), and slow (hours to days). The first two are believed to operate after moderate or intermittent stress; the third, in pathologic conditions or therapy with high glucocorticoid levels sustained for days.
Sensitivity to feedback depends on many factors, including the time of day. Basal ACTH release is less sensitive than stimulated release. Furthermore, a stressful stimulus in some way facilitates the ACTH response to a subsequent stress, overcoming the feedback inhibition due to the elevated glucocorticoid levels produced by the first stress. Some feedback can be seen as facilitative or permissive.72
Regulation of basal activity of the HPA axis requires glucocorticoid binding to both MRs and GRs. Inhibition of basal secretion of ACTH by corticosterone in rats at the low point of diurnal HPA activity (the morning) appears to occur through MRs, whereas inhibition at peak activity (evening) occurs through GRs potentiated by MRs.73 Suppression of stimulated ACTH secretion, which prevents overactivity in the stress-induced HPA axis, occurs through binding to GRs in pituitary corticotrophs and hypothalamic CRH neurons.
ACTH is produced as part of the larger precursor protein, POMC, which is also the progenitor of the melanocyte-stimulating hormones α- and β-MSH, β-endorphin, and the lipoproteins. Increased MSH activity associated with increased ACTH appears to be responsible for the changed skin color of Addisonian patients, as originally noted by Addison.2 Synthesis of POMC in pituitary corticotrophs is stimulated by CRH and is inhibited by glucocorticoids, at least partly at the level of transcription of the POMC gene. Direct repression by glucocorticoids occurs through nGREs, which may repress by disrupting interactions that maintain basal transcription. Indirect repression occurs via the hypothalamus.
Corticotrophs are also directly influenced by other hormones, including angiotensin II, paracrine secretions from neighboring pituitary cells, and cytokines such as TNF-α, IL-1, and IL-6.
HYPOTHALAMUS: CRH AND VP
Secretion of CRH and VP from the paraventricular nuclei, along with other ACTH secretagogues, is subject to both humoral and neural regulation. Secretion increases following adrenalectomy, is stimulated in a stress-specific manner by hemorrhage, injury, hypoglycemia, hypoxia, pain, fear, and other kinds of stress, and generally is inhibited by glucocorticoids74 (see Fig. 97-1). Some inhibition by glucocorticoids may occur via a nongenomic path involving rapid endocannabinoid release in the PVN.75 CRH output can be modulated by catecholamines, leptin, and several cytokines.76 Acute hemorrhage raises levels in hypothalamic neurons of mRNA for CRH but not VP. CRH, via CRH-1 receptors, is thought to orchestrate the immediate behavioral, sympathetic, and HPA axis responses to stress, whereas the CRH-related neuropeptides stresscopin and urocortins, which bind to CRH-2 receptors, may assist slower stress responses.58
In normal rats, stress activates CRH gene expression in the PVN, which is suppressed by glucocorticoids at high levels. In adrenalectomized rats, stress does not activate CRH gene expression unless the animals are first treated with glucocorticoids at low levels.77 The low, facilitative, or permissive levels are thought to act through MRs, and the high, suppressive levels through GRs.77
CRH knockouts homozygous for the defective gene are viable as long as they receive glucocorticoids during the period from a week before birth until 2 weeks after birth. Without glucocorticoids, they die within 12 hours of gestation owing to severe lung abnormalities, including low surfactant mRNA. Glucocorticoids are known to be important for lung development, particularly for synthesis of surfactant.78 Compared with normal mice, the CRH knockouts exhibited a drastically diminished rise in corticosterone levels in response to stress.79
In addition to controlling ACTH secretion, CRH has numerous actions within and outside the brain. When secreted by peripheral nerves, it acts as a proinflammatory agent: mRNAs for CRH-R1 and CRH-R2 are expressed in adipose tissue, whereas CRH downregulates 11β-HSD1.47,80 Stresscopin and urocortins, via CRH-2 receptors, reduce appetite and may participate in delayed stress responses.58
CYTOKINE FEEDBACK
As first proposed by Besedovsky and Sorkin,81 cytokines communicate between the immune system and the HPA axis. IL-1 has been shown to mediate HPA stimulation by endotoxin. IL-1α, IL-1β, IL-6, and TNF-α administered peripherally increase HPA activity with increased levels of glucocorticoids, ACTH, or POMC mRNA, and CRH or CRH mRNA. IL-1 causes release of both CRH and VP from neurosecretory cells.76 The brain has receptors for IL-1, IL-2, IL-6, and other cytokines, and it produces IL-1.82 (In Fig. 97-1, the question mark indicates uncertainty about which cytokines are most important and how their message is conveyed.)
Transgenic mice reveal the central role of IL-1 in feedback control and stress activation of the HPA axis. Mice lacking IL-1β fail to respond with increased plasma corticosterone to inflammatory stress, whereas mice lacking IL-1α respond normally, suggesting that IL-1β is crucial to the neuro-immuno-endocrine response.83 Mice lacking IL-1 receptor type 1 have diminished corticosterone responses to psychological, metabolic, and restraint stresses. These mice and mice with overexpressed IL-1 receptor antagonist targeted to the brain do not hypersecrete ACTH after adrenalectomy.66
Whether peripherally released cytokines like IL-1 enter the brain in physiologically significant amounts, and if not how their message reaches the hypothalamus, are unsettled issues. Among hypotheses that have been proposed are that cytokine messages are transmitted via the vagus or through specialized brain regions like the organum vascularis laminae terminalis (OVLT), or via mediators like eicosanoids, catecholamines, nitric oxide, or cytokines generated in the brain.
Physiologic Actions of Glucocorticoids
METABOLISM
Control of Blood Glucose
Glucocorticoids act in concert with other hormones to maintain or raise blood glucose levels by (1) stimulating hepatic gluconeogenesis, (2) mobilizing gluconeogenic substrates from peripheral tissues, (3) permissively enhancing and prolonging the effects of glucagon and epinephrine on gluconeogenesis and glycogenolysis, (4) inhibiting peripheral glucose utilization, and (5) promoting liver glycogen synthesis to store substrate in preparation for acute responses to glycogenolytic agents such as glucagon and epinephrine.45
From an evolutionary standpoint, glucocorticoids in this way support stress responses that require glucose for rapid and intense exertion, such as an encounter of prey with predator.26 From a physiologic and clinical standpoint, glucocorticoids are counterregulatory hormones that protect the body from insulin-induced hypoglycemia. Both of these roles, in which glucocorticoid effects develop over the course of hours, are shared with the rapidly acting glucagon and epinephrine and to some extent with growth hormone.84
Glucocorticoid actions interact with those of insulin during feeding and fasting in complex ways that not only maintain blood glucose but influence appetite, feeding patterns, disposal of foodstuffs, and body composition.26,85 Antagonism with insulin in both glucose synthesis and utilization accounts at least partly for the diabetogenic actions of excessive glucocorticoids.45,86 Studies with transgenic mice show that expression of hepatic peroxisome-proliferator–activated receptor-α (PPAR-α) may be one mechanism underlying glucocorticoid-induced hypertension and insulin resistance.87
Gluconeogenesis
Hepatic gluconeogenesis is stimulated by glucocorticoids, mainly through the increased activities of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase. These enzymes catalyze the conversion of oxaloacetate to phosphoenolpyruvate and of glucose-6-phosphate to glucose—both rate-limiting steps in gluconeogenesis.45,88 Glucocorticoids also regulate expression of 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase, a bifunctional enzyme that controls the level of fructose-2,6-biphosphate. Fructose-2,6-biphosphate is an allosteric regulator of gluconeogenic and glycolytic enzymes. PEPCK and 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase activities are controlled principally through synthesis of the enzymes.88 On starvation, 11β-HSD1 knockout mice have diminished activation of PEPCK and glucose-6-phosphatase.89
Control of PEPCK gene expression reflects the complexity of regulation of gluconeogenesis in the body, involving glucocorticoids, insulin, glucagon, catecholamines, cyclic adenosine monophosphate (cAMP), and retinoic acid.88,90 In particular, glucocorticoids and insulin, by respectively promoting and indirectly disrupting association of CBP (CREB-binding protein) and RNA polymerase II with the PEPCK promoter, reciprocally regulate PEPCK gene expression. The PEPCK gene has a glucocorticoid response unit (GRU) that spans 110 base pairs. There are two GR-binding sites and four accessory factor elements, all of which are required for glucocorticoid regulation, and within the GRU are insulin-responsive and retinoic acid–responsive sequences.88,91 The 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase gene has a complex glucocorticoid response element that resembles the GRU of the PEPCK gene. Hepatocyte nuclear factor-6 (HNF-6) inhibits glucocorticoid activation of both these genes by binding to DNA and GRs. As would be expected, treatment with glucocorticoids of transgenic mice with dimerization-deficient GRs (i.e., GRs that cannot bind to GREs) failed to induce PEPCK.35
Substrates for gluconeogenesis are generated by glucocorticoids through release of amino acids from muscle and other peripheral tissues and release of glycerol along with lipolysis.
Permissive actions of glucocorticoids on gluconeogenesis by glucagon and epinephrine, possibly due to enhanced responsiveness to cAMP or other intracellular mediators, are evidenced by the impairment of gluconeogenesis caused by adrenalectomy and its normalization by glucocorticoids.
Glucose Utilization
Glucocorticoid inhibition of peripheral glucose utilization can be demonstrated both in intact organisms and with isolated cells.92 It probably accounts for significant insulin antagonism and for the early rise in blood glucose seen after glucocorticoid treatment, and it may play a role in the release of gluconeogenic substrates from peripheral tissues. Glucose uptake is inhibited by direct glucocorticoid actions on normal skin, fibroblast, adipose tissue, adipocytes, lymphoid cells, and polymorphonuclear leukocytes. This inhibition, which requires RNA and protein synthesis, has been postulated to be mediated by a glucocorticoid-induced protein. It results mainly from translocation of glucose transporters from the plasma membrane to intracellular sites.93,94 Glucose uptake by muscle is inhibited in intact organisms treated with glucocorticoids. This action may be indirect.
Glycogen Synthesis and Breakdown
Stimulation of glycogen synthesis by glucocorticoids takes place in the fetus and in the adult. It depends on increased synthesis of hepatic glycogen synthase and activation by dephosphorylation of its inactive form, as well as on inactivation by dephosphorylation of phosphorylase a. Some of these changes can be accounted for by increases in glycogen-bound phosphatase activity.
Stimulation of liver glycogen synthesis by stress-induced glucocorticoids can be interpreted as preparation for a subsequent challenge in which glycogen will be used for rapid conversion to glucose by glycogenolysis.26 Furthermore, glucocorticoids at basal levels are required to permissively maintain epinephrine-induced glycogenolysis, which is impaired by adrenalectomy.
Fat Metabolism
Glucocorticoids, in opposition to insulin, inhibit glucose transport by adipose cells and stimulate free fatty acid release, which in humans results in an increase in plasma free fatty acids within 1 to 2 hours of administration of hormone.95 Increased release of fatty acids also occurs after incubation of adipose tissue with glucocorticoids, an effect that is due in part to decreased reesterification resulting from the decrease in glucose uptake, and in part to increased lipolysis. Stimulation of lipolysis is largely a permissive effect seen in the presence of growth hormone and other lipolytic agents.95 Glucocorticoids also inhibit the action of leptin and are permissive for the obesity syndrome in mutant rodents, which is ameliorated by adrenalectomy. A curious observation is that mice expressing a GR antisense construct that lowers their GR levels have as their most striking abnormality an increase in fat deposition, which can double their weight compared with normal mice. Because these mice eat less than normal, they are presumed to have increased energy efficiency.96
Chronic stress, which increases low diurnal concentrations of glucocorticoids, has been linked to central obesity and the metabolic syndrome, a disorder that combines diabetes, insulin resistance, dyslipidemia, and hypertension.61 11β-HSD1 knockout mice have weakened glucocorticoid-induced responses and resist hyperglycemia provoked by obesity or stress.89 The critical importance of local glucocorticoid levels has been demonstrated with transgenic mice that overexpress 11β-HSD1 in fat cells: those mice develop central obesity and the metabolic syndrome.45,47
Catabolic Effects
Chronic high levels of glucocorticoids lead to massive catabolic effects on proteins and other components of peripheral tissues, causing muscle wasting97 and lipolysis with redistribution of fat. These pathologic changes are probably magnified expressions of physiologic mechanisms for generating gluconeogenic substrates and may result from interactions with insulin and other hormones.45
BONE
Glucocorticoids act on bone and cartilage during development and adulthood. When present in excess, they cause osteoporosis and impair skeletal growth, inhibiting bone formation by decreasing the number of osteoblasts and their function, increasing collagenase expression, and inhibiting collagen synthesis.98 On osteoblasts, they exert both permissive and suppressive effects.99 At basal levels, they mobilize neutrophils from bone marrow to blood and other tissues100: possible molecular mechanisms of these glucocorticoid actions include decreased expression of insulin-like growth factor-1 (IGF-1), IGF-binding protein, IGF-1 and growth hormone receptors, and interactions with thyroid hormones. Glucocorticoids and cytokines in bone cells influence each other in complex ways, glucocorticoids generally suppressing cytokines and cytokines upregulating or downregulating GRs.101 Levels of glucocorticoids in bone appear to depend on local 11β-HSD1. CRH, through direct peripheral inflammatory effects rather than through glucocorticoids, induces in rats degeneration of cartilage and bone.102
IMMUNE AND INFLAMMATORY REACTIONS
Antiinflammatory and Immunosuppressive Actions
Among the major clinical applications of hormones is the use of glucocorticoids for suppression of inflammatory and immune reactions and for treatment of patients with cancers of the lymphoid system. (Evolution anticipated such a use with vaccinia virus, which encodes an enzyme, 3β-hydroxysteroid dehydrogenase, which in infected organisms enhances glucocorticoid production, suppressing the inflammatory response and increasing virulence.103)
As already described, for years glucocorticoid suppression of inflammatory and immune reactions was believed to result from pharmacologic actions with no physiologic significance. Strong evidence, however, points to their physiologic nature. They are elicited through the same receptor-mediated genomic mechanisms as physiologic effects. Adrenalectomy or administration of the glucocorticoid antagonist mifepristone (RU486) enhance responses to inflammatory agents, showing that endogenous glucocorticoids normally control inflammation and similarly control autoimmune reactions. A striking example is the high susceptibility to arthritis of Lewis rats compared to the largely histocompatible Fischer rats after challenge with streptococcal cell wall polysaccharide (SCW).104 This difference is due to a defect in biosynthesis of CRH that limits the glucocorticoid response of Lewis rats to a challenge. Lewis rats can be protected from SCW with dexamethasone, whereas Fischer rats become susceptible to SCW when pretreated with RU486. Defective HPA function may have an etiologic role in rheumatoid arthritis.105 In transgenic mice with GRs that cannot dimerize and therefore cannot transactivate genes through binding to palindromic GREs, most anti-inflammatory and immunosuppressive actions of glucocorticoids remain intact, indicating that they are mediated through binding of GRs to factors such as AP-1 and NF-κB.34
Glucocorticoids also exert permissive actions on the immune system, as will be described below. Stimulation of the HPA axis may mobilize immunoregulatory agents other than glucocorticoids. One already mentioned is CRH, which peripherally has proinflammatory activity.104 Whether such agents normally participate in immune responses to stress is uncertain.
Effects on Leukocytes
Glucocorticoids influence most cells responsible for immune and inflammatory reactions, including lymphocytes, natural killer (NK) cells, monocytes and macrophages, dendritic cells, eosinophils, neutrophils, mast cells, and basophils. Accumulation of most of these cells is decreased at inflammatory sites, an effect that can be induced by local application of hormones, but they mobilize neutrophils from bone marrow to blood.100,106 Glucocorticoids may have both beneficial and detrimental effects on wound healing.107 Blood counts of lymphocytes, monocytes, eosinophils, and basophils drop within 1 to 3 hours of glucocorticoid administration, generally recovering in 12 to 48 hours. NK cells are unaffected, and neutrophil counts rise. CD4 or helper T cells are more sensitive to lymphopenia than are B cells, and CD8 or cytotoxic T cells are relatively insensitive. Increased neutrophil number is thought to reflect increased release of marginated cells to the circulation and increased half-life. These alterations in cell traffic probably depend on inhibiting expression of surface molecules such as endothelial leukocyte adhesion molecule (ELAM)-1 and intercellular adhesion molecule (ICAM)-1,104,108 thus decreasing adhesion of leukocytes to endothelial and other cells.
Glucocorticoid administration usually reduces antigen- or lectin-induced mitogenesis measured with peripheral lymphocytes, an effect also observed with lymphocytes in culture. T cells are more sensitive than B cells, and helper more sensitive than cytotoxic T cells. Glucocorticoids also directly inhibit T and B cell proliferation, early B cell differentiation, NK activity, and the differentiation and function of macrophages. They inhibit antigen presentation by monocytes and by dendritic cells (the most potent antigen-presenting cells) and shift responses from T helper 1 (Th1) cells to Th2 cells109 Although glucocorticoids have stimulatory effects on immunoglobulin synthesis in cell culture, in whole organisms glucocorticoids usually inhibit B cell function.
Permissive glucocorticoid actions on T cell function have been observed in human volunteers treated with lipopolysaccharide (LPS), a mediator of septic shock: when administered within 6 hours of LPS, cortisol hemisuccinate suppressed the LPS-induced increase in TNF, but when given 12 to 144 hours before LPS, it magnified the TNF response.110 Both in rats and in cultured splenic lymphocytes, glucocorticoids at low concentrations, presumably acting through MRs, can enhance T cell responses to concanavalin A, whereas at higher concentrations, through GRs, they suppress.111 Hormone concentration and timing appear to be important for these actions to be displayed separately from the usually predominant suppressive effects.112 Delayed-type hypersensitivity (DTH) reactions to cutaneous antigen exposure are enhanced by acute physiologic increases in glucocorticoid levels (and eliminated by adrenalectomy), but they are suppressed by chronic exposure to glucocorticoids.113 In treatment for contact hypersensitivity, the therapeutic action of glucocorticoids is exerted through macrophages and neutrophils and requires GR dimerization.114 The value of glucocorticoids in treatment for septic shock is controversial108 (see Chapter 115).
Important effects of glucocorticoids are exerted through the innate immune system. This ancient defense system uses about 10 invariant germline-encoded toll-like receptors (TLRs) on monocytes, macrophages, and dendritic and other cells to respond to infectious molecules of microbial origin by inducing antimicrobial genes along with inflammatory cytokines and chemokines, thus stimulating leukocyte migration and triggering adaptive immune responses.115 LPS, the best-known stimulator of the innate system, provokes a rapid increase in glucocorticoid levels, which, in turn, protects the organism from potentially lethal effects of LPS.108,116 The HPA response and other effects of LPS are mediated in part by the proinflammatory cytokines TNF-α, IL-1, and IL-6. In the airway epithelium, glucocorticoids may enhance innate immunity.117,118 Glucocorticoid treatment of myeloid progenitors also enhances TLR signaling in macrophages to which they differentiate.119 In other circumstances, glucocorticoids inhibit TLR signaling.120
Glucocorticoids protect against overactivity of immune reactions through several mechanisms, including suppression of production or potentially toxic activity of proinflammatory cytokines, histamine, adhesion molecules, inducible cyclooxygenase, and inducible nitric oxide synthase. Glucocorticoids also suppress expression and release of the LPS receptor CD-14. Overexpression of GRs increases resistance to LPS, thereby reducing production of IL-6.121
The anti-inflammatory cytokine IL-10 may play an important role in controlling LPS effects. Neutralization of IL-10 enhances the lethality of LPS in mice, whereas IL-10 administration reduces lethality.122 Optimal IL-10 production during cardiac surgery requires a surge in glucocorticoid levels.123 In experimental human endotoxemia, even high doses of glucocorticoids enhance IL-10 production.124
During development, LPS may affect the HPA axis and have long-lasting effects on immune regulation. Exposure of neonatal rats to LPS raises their adult corticosterone levels and protects the adults from adjuvant-induced arthritis.125
Apoptosis
Glucocorticoid-induced apoptosis of thymocytes and other lymphocytes is among the most striking effects of these hormones,12,126,127 and the underlying mechanisms of this effect are being revealed gradually.31 Apoptosis is directly linked to control of T cell pools in vivo, as is demonstrated with transgenic mice. Mice with increased GR expression have increased sensitivity to glucocorticoid-induced apoptosis,121 and mice with higher or lower GR levels targeted to lymphocytes have, respectively, smaller or larger T cell pools.128 Glucocorticoid-induced apoptosis has been demonstrated with most hematologic cells, as well as with other cells such as epithelial and carcinoma cells and osteoblasts.129,130 In some circumstances, glucocorticoids protect thymocytes from apoptosis69; such antiapoptosis is also found with neural and other cells.129–133
The physiologic significance of these effects is uncertain. Glucocorticoid-induced apoptosis has been invoked to account for immunosuppression, which, at least for short-term effects (hours or days), is better explained by actions on cytokines as described below. Apoptosis might serve to eliminate toxic or otherwise dangerous activated lymphocytes.134 Several plausible ideas have been proposed for glucocorticoid involvement in positive or negative thymic selection of the T cell repertoire.69 Thymocyte apoptosis, in contrast to many antiinflammatory and immunosuppressive reactions of glucocorticoids, requires GRs that dimerize (i.e., it is mediated by transactivation of genes through palindromic GREs).34
Effects via Cytokines and Other Mediators, and via Their Receptors
Many suppressive effects of glucocorticoids on immune and inflammatory reactions appear to be due to inhibition of production or activity of cytokines, chemokines, inflammatory agents, certain hormones and neurotransmitters, and other mediators that are released during responses to LPS and other forms of stress.108,135,136 Although many of these results were originally obtained with cell cultures, most have been observed in intact organisms.26
These mediators form communication networks for defense mechanisms that respond to stress-induced challenges to homeostasis: cytokines and chemokines respond to infection, inflammatory agents to tissue damage, neurotransmitters to “fight or flight” encounters, and so forth. By blocking communication, glucocorticoids limit the stress response, preventing it from overshooting and damaging the organism. Most mediators in excess can be toxic, even lethal. Glucocorticoids limit not only production of mediators but sometimes their effects, such as TNF-α toxicity and responses of lymphocytes to IL-2 and of eosinophils to IL-3, IL-5, interferon (IFN)-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF).137
Chemokines or chemotactic cytokines are produced locally in tissues, thus influencing traffic and homing of leukocytes by binding to G protein–coupled cell-surface receptors. Their secretion, probably stimulated by such cytokines as IL-1 and TNF-α, is dramatically increased during inflammation, resulting in recruitment of leukocytes to the inflamed site. Significant glucocorticoid effects on cell traffic are due to inhibition of chemokine secretion and cell adhesion molecules.
Not all mediators are suppressed by glucocorticoids. Annexin 1 (lipocortin 1) is induced by glucocorticoids.138 It is antiinflammatory in several systems and may mediate inhibitory effects of glucocorticoids on release of ACTH from the pituitary.139 Macrophage migration inhibitory factor (MIF) represents a special case because it antagonizes glucocorticoid actions.140 In vivo, glucocorticoids raise MIF levels in plasma and in thymus, spleen, and other cells, and MIF in turn counteracts glucocorticoid effects.136,141 Some mediators, like IL-10, are stimulated under some conditions and are suppressed under others. In the acute phase response, which involves the proinflammatory cytokines IL-1, IL-6, and TNF-α, glucocorticoids both potentiate induction by cytokines of certain acute phase proteins and suppress production of the cytokines.142
Several mediators that are suppressed by glucocorticoids have receptors that, paradoxically, are induced by glucocorticoids. As discussed below, glucocorticoid induction of mediator receptors is a possible mechanism for some permissive effects.
Molecular mechanisms by which glucocorticoids control mediator production vary.108,143 IL-1 production is blocked at the levels of transcription, translation, and secretion. TNF-α and GM-CSF appear to be blocked through increased degradation of their mRNAs. IL-2, IL-3, and possibly IFN-γ are blocked at the transcriptional level. Some, like prostaglandins and nitric oxide, may be suppressed because induction of the enzyme that synthesizes them is inhibited. Underlying many of these effects may be GR protein-protein cross-talk with NF-κB, AP-1, and other transcription factors,143 which is consistent with the fact that most antiinflammatory actions are unaffected in transgenic mice with GRs that cannot dimerize.34 Mitogen-activated protein kinase (MAPK) phosphatase 1 (MKP-1) is involved in MIF-glucocorticoid cross-talk.141 Numerous other mechanisms are being investigated.
Not only do glucocorticoids influence cytokines, but glucocorticoid actions are regulated by such cytokines as MIF, which was already mentioned, and IL-1, IL-2, TNF-α, IL-10, and IL-11.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


