FIGURE 61-1. Nomogram for calculating TmP/GFR. Calculations are done from fasting samples obtained in the morning. The urine is collected for 1 to 2 hours (the time period is not critical). The CPO4/Ccreat = (UPO4 × [Creat])/(Ucreat × [Pi]). To use the nomogram to calculate TMP/GFR, a straight line is passed through the appropriate plasma phosphate concentration and the CPO4/Ccreat. The line will intersect with the corresponding TMP/GFR value. For SI units, use the inside scales. For metric (mg/dL), use the outside scales.
(From Walton RJ, Bijvoet OL: Nomogram for derivation of renal threshold phosphate concentration. Lancet 2:309–310, 1975.)
REGULATION OF PHOSPHATE REABSORPTION
Parathyroid Hormone
PTH has a major effect on serum phosphate. The clinical relevance of this effect is demonstrated in patients with hyperparathyroidism, who develop phosphaturia and hypophosphatemia, as well as those with hypoparathyroidism, who have increased phosphate reabsorption and hyperphosphatemia. PTH rapidly (15 to 60 minutes) reduces the number of NPT2a cotransporters on the apical surface of the cells in the renal proximal tubule. The effect appears to result from microtubule-dependent internalization into endocytic vesicles and subsequent destruction of the transporters.73,74 This acute down-regulation of NPT2a cotransporters does not involve reduction in NPT2a gene transcription,75 although transcriptional suppression is seen after more prolonged PTH exposure.76 After parathyroidectomy, rats manifest a two- to threefold increase in both protein and messenger RNA (mRNA) levels of NPT2a, which correlates with a striking increase in phosphate reabsorption.75 Evidence exists for involvement of both PKA and protein kinase C (PKC), as well as MAPK signaling via PTH/PTHrP receptors in bringing about these responses.77,78 Despite the fact that PTH clearly has a major effect on phosphate homeostasis, it should be kept in mind that the primary role of PTH is to regulate serum calcium level and not phosphate homeostasis. Recent studies of the role of FGF23 further illuminate this issue (see later).
Dietary Phosphate
Renal phosphate excretion is extremely sensitive to changes in dietary phosphate availability. Thus, dietary phosphate deprivation79–86 or supplementation80,82,87 rapidly evokes a compensatory increase or decrease, respectively, in renal phosphate reabsorption ([Pi]Th). Compelling clinical and experimental evidence has established that these adaptations to dietary phosphate occur quite independently of PTH.74,75,79–82,88
Increased sodium-dependent phosphate transport by isolated brush-border membrane vesicles occurs within a few hours of phosphate deprivation86,89 and reflects increased maximal velocity (rather than affinity) of the phosphate carrier,89–91 consistent with an increased number of membrane transporters. This has been corroborated by direct immunohistologic demonstration that institution of a low-phosphate diet causes rapid (within 2 hours) insertion of NPT2a cotransporters into the apical plasma membrane of rat proximal tubular cells by a microtubule-dependent mechanism.92–94 Up-regulation of NPT2a gene transcription occurs subsequently during more prolonged phosphate restriction (i.e., several days).74 Similarly, high dietary phosphate rapidly reduces apical membrane NPT2a protein levels, with no change in NPT2a gene transcription for at least several hours.74,95
Fibroblast Growth Factor-23
Fibroblast growth factor-23 (FGF23), the gene identified as causative for autosomal dominant hypophosphatemic rickets (see later) and some forms of familial hyperphosphatemic tumoral calcinosis, does not appear to play a role in the rapid changes in phosphate reabsorption due to acute changes in intestinal phosphate.96 However, there is substantial evidence that FGF23 plays a role in maintenance of normal phosphate homeostasis over the course of days.
FIBROBLAST GROWTH FACTOR-23 ACTIVITY
FGF23 has similar functions as PTH to reduce renal Pi reabsorption but has opposite effects on 1,25(OH)2D production. FGF23 delivery leads to renal Pi wasting through the down-regulation of both NPT2a and NPT2c.97 Under normal circumstances, hypophosphatemia is a strong positive stimulator for increasing serum 1,25(OH)2D production. However, patients with ADHR, TIO, XLH, and ARHR manifest hypophosphatemia with paradoxically low or inappropriately normal serum 1,25(OH)2D concentrations. In mice, the expression of the 1α(OH)ase enzyme and the catabolic 24(OH)ase are reduced and elevated, respectively, when the animals are exposed to FGF23 by injection or by transgenic approaches.98 Thus, the effects of FGF23 on the renal vitamin D metabolic enzymes is most likely responsible for the reductions in 1,25(OH)2D in the setting of often marked hypophosphatemia in ADHR, XLH, TIO, and ARHR patients.
REGULATION OF FGF23 PRODUCTION
In humans, dietary Pi supplementation increased FGF23, whereas Pi restriction and the addition of Pi binders suppressed serum FGF23 (Fig. 61-2),99 indicating that FGF23 plays a role in maintenance of Pi homeostasis. In animal studies, the FGF23 response to serum Pi appears to be much more dramatic than in the human studies. Mice given high and low Pi diets have increased and decreased serum FGF23 levels, respectively.100
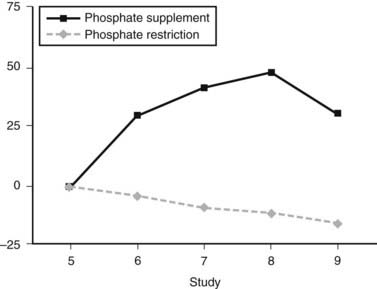
FIGURE 61-2. FGF23 levels in response to diet. Percent change in FGF23 during the intervention period (♦, phosphate depletion intervention as measured with the intact FGF23 assay;  , phosphate loading intervention as measured with the intact FGF23 assay). The intervention, dietary phosphate depletion, or loading was started after the day 5 samples were collected. Values are presented as mean ± SE.
, phosphate loading intervention as measured with the intact FGF23 assay). The intervention, dietary phosphate depletion, or loading was started after the day 5 samples were collected. Values are presented as mean ± SE.
(Data from Burnett SM, Gunawardene SC, Bringhurst FR et al: Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women, J Bone Miner Res 21:1187–1196, 2006.)
Vitamin D has important regulatory effects on FGF23 in vivo. In mice, injections of 20 to 200 ng 1,25(OH)2D led to dose-dependent increases in serum FGF23 concentrations.98 These changes in FGF23 occurred before detectable changes in serum Pi, indicating that FGF23 may be directly regulated by vitamin D. Physiologically, this would be consistent with results examining the role of FGF23 in vitamin D metabolism, in that FGF23 has been shown to down-regulate the 1α(OH)ase mRNA.97,98 Thus as 1,25(OH)2D is elevated in the blood as a product of 1α(OH)ase activity, vitamin D would then increase FGF23 production, which would complete the negative-feedback loop and down-regulate 1α(OH)ase.
Serum Assays
FGF23 can be measured in the bloodstream via several assays. One widely used assay is a “C-terminal” FGF23 enzyme-linked immunosorbent assay (ELISA), with both the capture and detection antibodies binding C-terminal to the FGF23 176RXXR179/S cleavage site.101 This assay thus recognizes full-length FGF23 as well as C-terminal fragments that could arise through proteolytic processing. The C-terminal assay is quantified relative to standards composed of FGF23-conditioned media produced from stable cell lines expressing the human protein, and it only recognizes the human FGF23 isoform. The normal mean for this assay is 55 ± 50 reference units (RU)/mL, and the upper limit of normal is 150 RU/mL. In a study with a large number of controls and TIO patients, this ELISA was used to examine FGF23 concentrations in TIO and XLH101 and showed that serum FGF23 is detectable in normal individuals. The mean FGF23 was greater than 10-fold elevated in TIO patients, which rapidly fell after surgical removal of the tumor. Importantly, most XLH patients (13 out of 21) had elevated FGF23 compared to controls,101 and in those with “normal” FGF23, these levels may be “inappropriately normal” in the setting of hypophosphatemia.
An “intact” FGF23 assay has been developed that uses conformation-specific monoclonal antibodies that span the 176RXXR179/S180 SPC cleavage site (see later) and thus recognize N-terminal and C-terminal regions of the FGF23 polypeptide.102 In normal individuals, this assay has a mean circulating concentration of 29 pg/mL. The published upper limit of normal is 54 pg/mL.102 The results of these two assays generally agree with regard to the relative ranges of FGF23 concentrations in XLH and in TIO patients, and that FGF23 is elevated in most XLH patients. Based upon limited data from two TIO patients undergoing resection, the intact assay was used to determine that the half-life of FGF23 is between 20 and 50 minutes.103,104
FGF23-Associated Syndromes
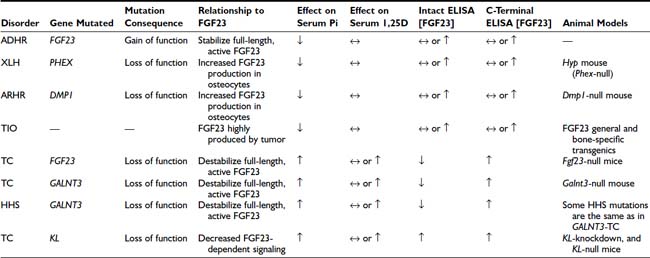
FGF23-associated syndromes, summarized in Table 61-1, can be divided into three groups:
DISORDERS ASSOCIATED WITH INCREASED FGF23 BIOACTIVITY
Autosomal-Dominant Hypophosphatemic Rickets (OMIM No. 193100)
ADHR is a rare disorder characterized by low serum Pi concentrations due to decreased TmP/GFR and inappropriately low or normal circulating vitamin D concentrations.105 ADHR was first described in a small family,106 and subsequently, a large ADHR kindred with many affected individuals was evaluated.105 This kindred provided an opportunity to test the phenotypic variability of ADHR in a large number of individuals with the same mutation. There was no evidence of genetic anticipation or imprinting. In contrast to the other genetic renal phosphate-wasting disorders, ADHR displays incomplete penetrance and variable age of onset. Important to the diagnosis and clinical management of ADHR, it was observed that this expanded ADHR family contains two subgroups of affected individuals. One subgroup consists of patients who presented during childhood with Pi wasting, rickets, and lower-extremity deformity in a pattern similar to the classic presentation of XLH. The second group consists of individuals who presented clinically during adolescence or adulthood. These individuals had bone pain, weakness, and insufficiency fractures but did not have lower extremity deformities.105 The patients with adult-onset ADHR had clinical presentations essentially identical to patients who present with TIO (none of the ADHR patients developed tumors). The molecular mechanisms for early-onset ADHR resembling XLH and late-onset ADHR resembling TIO are currently unknown. To date, all patients that have been described with delayed onset of clinically evident disease are female. In addition to these two groups, we found unaffected individuals who are carriers for the ADHR mutation and two patients who were treated for hypophosphatemia and rickets but later lost the Pi wasting defect.105 Thus, the clinical manifestations of ADHR are more variable than those observed in XLH.
To identify the gene for ADHR, the ADHR Consortium undertook a family-based positional cloning approach. A genomewide linkage scan in the large ADHR kindred described earlier demonstrated linkage to chromosome 12p13.3 (homologous to mouse chromosome 6).107 FGF23 was identified using exon prediction programs on genomic DNA sequence from the Human Genome Project.108 The FGF23 gene is composed of three coding exons and contains an open reading frame of 251 residues.108 The tissue with the highest FGF23 expression is bone, where FGF23 mRNA is observed in osteoblasts, osteocytes, flattened bone-lining cells, and osteoprogenitor cells.109 Quantitative RT-PCR showed that FGF23 mRNA was most highly expressed in long bone, followed by thymus, brain, and heart.110
Western blot analysis has demonstrated that wild-type FGF23 is secreted as a full-length 32-kD protein, as well as cleavage products of 20 kD (N-terminal) and 12 kD (C-terminal).110–112 Cleavage of FGF23 occurs within a subtilisin-like proprotein convertase (SPC) proteolytic site (176RXXR179/S180) that separates the conserved FGF-like N-terminal domain from the variable C-terminal tail (Fig. 61-3).
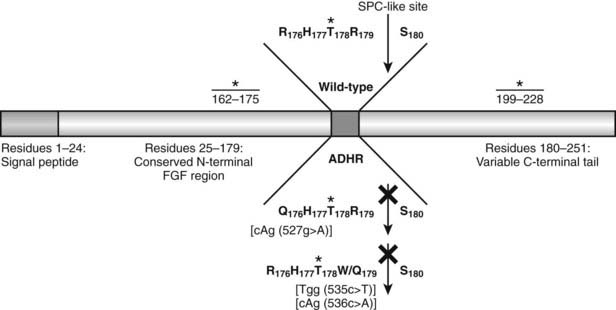
FIGURE 61-3. Model of FGF23 protein domains and effect of the ADHR mutations. FGF23 has a 24-residue signal peptide followed by residues 25 to 179 that comprise the conserved N-terminal FGF-like domain. The SPC-like site is interrupted by the ADHR mutations at R176 and R179 and divides the FGF-like domain from the variable C-terminal tail region. FGF23 undergoes glycosylation at three regions (denoted *), with the T178 most likely being the glycosylated residue that protects the mature protein from SPC degradation between R179 and S180 and is therefore critical for maintaining intact, active FGF23.
The ADHR mutations replace arginine (R) residues at FGF23 positions 176 or 179 with glutamine (Q) or tryptophan (W) within the SPC cleavage site, 176RXXR179/S180 (Table 61-2 and see Fig. 61-3).108,111,112 The SPCs are a family of serine proteases that process a wide variety of proteins including neuropeptides, peptide hormones, growth factors, membrane-bound receptors, blood coagulation factors, and plasma proteins.113 SPC substrates are cleaved C-terminal to the basic motif K/R-Xn-K/R, where Xn = 2, 4, or 6 residues of any amino acid.114,115 The SPCs, such as the furin protease, are largely expressed in the trans-Golgi network and possess similar but not exact substrate specificities. Following insertion of these mutations into wild-type FGF23, FGF23 secreted from mammalian cells was primarily produced as the full-length protein (32 kD) active polypeptide, as opposed to the 32-kD cleavage products typically observed for wild-type FGF23 expression.111 Peptide sequencing demonstrated that the 32-kD FGF23 form corresponded to full-length FGF23 after cleavage of the signal peptide (residues 25 to 251) and that the 12-kD isoform was the C-terminal portion of FGF23 downstream from the SPC cleavage site after R179 (residues 180 to 251) (see Fig. 61-3).112 As further evidence that FGF23 is processed intracellularly, the cleavage of wild-type FGF23 between R179/S180 is inhibited by a nonspecific SPC competitive inhibitor, Dec-RVKR-CMK, at concentrations between 25 and 50 µM.110,116 These studies show that the RXXR motif in FGF23 is central to its intracellular processing.
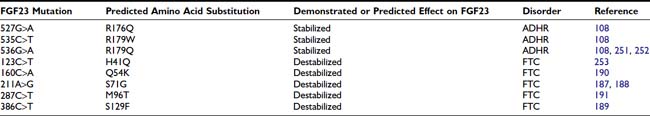
The SPC family is usually associated with the production of the active form of their substrate polypeptides. However, cleavage of FGF23 at the RXXR motif appears to be inactivating. In this regard, when full-length FGF23 or N-terminal (residues 25 to 179) and C-terminal (residues 180 to 251) fragments were injected into rodents, only the full-length protein lowered circulating phosphate concentrations.112 Since the full-length form of FGF23 induces hypophosphatemia, it is likely that the ADHR mutations increase the biological activity of FGF23 by stabilizing the full-length form and increasing its concentrations in the serum. Indeed, severely affected ADHR patients have increased circulating levels of FGF23.117
Tumor-Induced Osteomalacia
TIO is an acquired disorder of renal Pi wasting that is associated with tumors. Patients with TIO present with hypophosphatemia with inappropriately suppressed 1,25(OH)2D concentrations and elevated alkaline phosphatase levels.118 Osteomalacia is observed in bone biopsies. Clinical symptoms include gradual onset of muscle weakness, fatigue, and bone pain, especially from ankles, legs, hips, and back.118,119 Insufficiency fractures are common, and proximal muscle weakness can become severe enough for patients to require a wheelchair or become bed bound.118
The study of TIO introduced the idea for the existence of possible tumor-produced circulating factors, referred to as phosphatonins, that act upon the kidney to decrease Pi reabsorption.120,121 Support for these factors primarily comes from the knowledge that if the responsible tumor is surgically removed, the abnormalities in Pi wasting and vitamin D metabolism are rapidly corrected, as well as the fact that PTH, which decreases renal Pi reabsorption, is usually within normal ranges in TIO patients. Other studies have supported this hypothesis by showing that implantation of tumor tissue into nude mice resulted in increased urinary Pi excretion.122 To determine whether FGF23 could be involved in TIO as phosphatonin, five TIO tumors and several control tissues were tested by Northern blot for the presence of FGF23 transcripts, and it was determined that FGF23 mRNA was highly expressed in all of these tumors.123 Furthermore, FGF23 was present in a tumor lysate by Western blot analysis, with an anti–human FGF23 antibody.123
FGF23 was also independently identified in RNA from TIO tumors. Transcripts from tumors were isolated by differential selection using comparisons to mRNAs present in normal bone.124 The highly expressed transcripts were then subcloned, and the individual mRNAs were stably expressed in Chinese hamster ovary (CHO) cells, then injected into nude mice to form tumor masses.124 The cells that produced FGF23 recapitulated the TIO phenotype in vivo by causing hypophosphatemia, elevated alkaline phosphatase, and inappropriately low 1,25(OH)2D concentrations.124 In addition, the mice that received implanted cells also showed growth retardation, kyphosis, and osteomalacia. Further, there was marked decrease in the renal 1α(OH)ase. Both the biochemical and metabolic bone profiles were remarkably similar to those observed in TIO and ADHR patients. These experiments provided evidence that FGF23 was a phosphaturic substance and had dramatic effects on enzymes involved in vitamin D metabolism, and increased circulating FGF23 concentrations were consistent with the idea that FGF23 was at least in part responsible for the TIO phenotype. Serum FGF23 is elevated in patients with TIO,101,102 and tumors that cause TIO have a dramatic overexpression of FGF23 mRNA.123 Surgical resection of the tumor results in rapid decreases in serum FGF23.101
X-Linked Hypophosphatemic Rickets (OMIM No. 307800)
XLH is an X-linked dominant disorder and the most common form of heritable rickets.125 XLH is fully penetrant with variable severity. XLH patients present with laboratory findings that include hypophosphatemia with normocalcemia and inappropriately normal or low 1,25(OH)2D concentrations.125 Skeletal defects include lower-extremity deformities from rickets, bone pain, osteomalacia, fracture, and enthesopathy (calcification of the tendons and ligaments).125 It was determined by the Hyp Consortium that XLH is caused by inactivating mutations in PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome).126 Based upon sequence homology, PHEX encodes a protein that is a member of the M13 family of membrane-bound metalloproteases. Other members of this enzyme class include neutral endopeptidase (NEP) and endothelin-converting enzymes 1 and 2 (ECE-1 and ECE-2).126,127 This protease family is known to cleave small peptide hormones, therefore it is likely that PHEX has similar activity. Over 160 inactivating PHEX mutations have been described in XLH patients, including genomic variations that cause missense, nonsense, frameshift, and splicing changes (see PHEX Locus database: www.phexdb.mcgill.ca). Of note, although XLH is a renal Pi wasting disorder, PHEX shows the highest expression in bone cells such as osteoblasts, osteocytes, and odontoblasts in teeth, as well as lower expression in the parathyroid glands, lung, brain, and skeletal muscle but no expression in kidney.128 Taken together with the biochemical phenotype of XLH, PHEX protein homology and tissue expression are consistent with the hypothesis that PHEX interacts with small circulating factors outside of the kidney.
A valuable tool for the study of the pathophysiology of XLH has been the Hyp mouse, which has 3′ deletion in the Phex gene from intron 15 through the 3′ UTR129 and does not make a stable Phex transcript.128 This rodent model parallels the XLH phenotype, characterized by hypophosphatemia with inappropriately normal 1,25(OH)2D levels and normal serum calcium, as well as growth retardation and bone mineralization defects.130 Parabiosis studies between the Hyp mouse and a normal mouse pointed to the presence of a humoral factor, a phosphatonin, being transferred through the circulation of the Hyp mouse to the normal mouse to cause isolated renal Pi wasting.131 After the identification of PHEX/Phex, it was logically postulated that the enzyme may directly degrade a phosphaturic substance; however, recent studies suggest a more complex pathophysiology.
X-Linked Hypophosphatemic Rickets and Fibroblast Growth Factor-23
As described earlier, patients with XLH have overlapping phenotypes with ADHR patients. Because XLH results from a mutation in PHEX, which shares homology to a family of extracellular proteases, and ADHR arises from mutations in a protease cleavage site, it was logically hypothesized that circulating FGF23 would be cleaved and inactivated by PHEX. Thus, by mutational inactivation of PHEX in XLH, serum FGF23 concentrations would then elevate and cause renal Pi wasting. As described earlier, lending further support to this hypothesis were parabiosis studies between Hyp and normal mice, which pointed to the presence of a humoral phosphaturic factor in the Hyp mouse being transferred to the normal mouse. However, evidence to date has not supported a direct enzyme-substrate relationship between FGF23 and PHEX. In this regard, it was shown that recombinant PHEX did not cleave FGF23 but did cleave a positive control substrate.110 Furthermore, another report provided evidence that recombinant FGF23 was not cleaved by PHEX in cultured HEK293 cells coexpressing the proteins.116 This latter study expressed native FGF23 that was not epitope tagged to ensure that the additional residues did not cause conformational changes within FGF23 and interfere with potential PHEX activity.116
Several reports have established that FGF23 is elevated in many XLH patients.101,102,132 To understand the possible relationship between PHEX and FGF23, quantitative real-time RT-PCR was used to test Hyp bone for FGF23 mRNA concentrations versus wild-type bone. Interestingly, FGF23 mRNA in bone tissue from Hyp mice was elevated compared to levels present in control mice,110 and serum concentrations of FGF23 have been reported to be 10-fold higher in Hyp mice when compared to normal mice (our unpublished results and Ref. 133). This finding provides support for the idea that there is a cellular connection between inactive PHEX mutants (or lack of Phex expression in Hyp mice) and the up-regulation of FGF23 mRNA in bone cells. The elevated FGF23 mRNA levels may indicate that the increase in serum FGF23 in XLH patients is due to overproduction and secretion of FGF23 by skeletal cells, as opposed to the alternative hypothesis of a decreased rate of FGF23 degradation by cell surface proteases after secretion into the circulation. Although the interactions between FGF23 and PHEX are most likely indirect, the encoded proteins are coexpressed in osteoblasts and osteocytes.109,110,128 At present, the PHEX substrate and the mechanisms for phosphate sensing are unknown.
The current therapy for XLH, ADHR, and TIO includes oral replacement of phosphorus in combination with high-dose 1,25(OH)2D. This regimen “treats” XLH by increasing serum Pi concentrations and ameliorates much of the metabolic bone disease, but it does not directly “cure” the disorder by reversing the underlying molecular defects in kidney and in bone. In this regard, several studies have attempted to reverse the XLH phenotype. Transgenic expression of wild-type PHEX under the control of the bone-specific mouse pro-alpha(I) collagen gene134 and the osteocalcin (OG2)135 promoters on the Hyp background was undertaken. Interestingly, the defective mineralization of bone and teeth in the Hyp mice was partially resolved with PHEX under the regulation of the collagen promoter, and dry ash weight increased with the OG2 PHEX, indicating improved mineralization. However, the hypophosphatemia was not normalized in either study, indicating that expression of PHEX under the temporal regulation of an osteoblast-specific promoter is not sufficient to rescue the Hyp phenotype. Furthermore, expression of PHEX to levels observed in wild-type animals was not obtained in all studies. Importantly, a recent report of a transgenic model overexpressing PHEX in the Hyp mouse—using the human beta-actin promoter for directing expression in a wider tissue distribution (bone, skin, lung, muscle, heart)—resulted in similar results as the bone-specific promoter studies,136 further demonstrating that proper spatial-temporal expression of Phex is critical for normal mineral metabolism.
Treatment of X-Linked Hypophosphatemic Rickets
The current standard of care is combination therapy with phosphate and 1,25(OH)2D (calcitriol) or 1-hydroxyvitamin D3.137–139 Therapy is labor intensive, and patients and caregivers should understand this before initiating treatment. As in any medical encounter, patients should be fully informed about potential side effects of therapy.
The objectives of therapy in children are to correct or prevent deformity from rickets, promote growth, and lessen bone pain. In some cases, marked improvement in lower-extremity deformity is noted with medical therapy. In this regard, we frequently give a course of medical therapy before performing osteotomies to correct lower-extremity deformities. Most affected children probably should be treated if a system exists to administer and monitor therapy. However, in some instances, it may be acceptable not to treat but rather to follow mildly affected children carefully (no less than twice a year).
The indications for treatment of adult patients are more controversial. There are no data to suggest that current treatment regimens prevent enthesopathy (calcifications of tendons and ligaments). Pseudofractures are common in moderately to severely affected adult XLH patients. Since pseudofractures are often painful, may lead to fracture, and generally respond well to treatment, we generally recommend medical therapy for patients with pseudofractures. XLH patients frequently complain of bone pain,121 presumably due to osteomalacia. Treatment lessens osteomalacia and bone pain, and it is therefore reasonable to treat patients with this complaint. Additionally, it is advisable to treat patients who have nonunion after fractures or osteotomies, because treatment may improve fracture healing. In light of the complexity of therapy, potential side effects (see later), and lack of increased risk of fracture in patients without pseudofractures,121 therapy is not recommended in asymptomatic patients who do not have pseudofractures.
Once the decision is made to initiate therapy, it is best to start at a low dose of calcitriol and phosphate (to avoid diarrhea from phosphate) and gradually increase therapy over several months. Some clinicians maintain a “high dose” phase for up to 1 year. During this phase of therapy, the calcitriol dose may be as high as 50 ng/kg per day in two divided doses, but not more than 3.0 mcg/day. However, not all experienced clinicians use this high-dose phase and some will use a maximum of 25 ng/kg. Also, 20 to 40 mg/kg of phosphate is administered in four divided doses (up to a maximum of 2 g per day). Serum calcium, phosphorus, and creatinine levels, as well as urine calcium and creatinine concentrations, are routinely monitored on a monthly basis during the high-dose phase. The doses of calcitriol and phosphate are adjusted based on the laboratory results. Hypercalciuria may be the first sign of healing of osteomalacia (theoretically, osteomalacic bone is capable of taking up more calcium and phosphate than bone that is histologically normal). Although some clinicians recommend administering phosphate over five doses per day initially, we do not ask patients to wake up to take medication at night for the following reasons: (1) serum phosphate concentrations tend to rise at night in XLH patients,140 (2) there is an exaggerated nocturnal rise in PTH concentrations in XLH patients, and (3) it is very difficult for patients to wake up to take medication every night for a chronic condition. After approximately 1 year on high-dose therapy, patients are switched to a long-term “maintenance” phase with approximately 10 to 20 ng/kg per day of calcitriol and no change in the dose of phosphate. While patients are on maintenance therapy, we continue to see them and monitor serum and urine biochemistries at least every 3 to 4 months.
Preliminary data indicate that standard therapy may have the undesirable affect of increasing FGF23 concentrations. It is possible that calcitriol, phosphate, or both stimulate the production of FGF23 in XLH patients despite the fact that they are still hypophosphatemic.141 This is currently an active area of research; however, physicians may want to consider not using a high-dose phase.
Careful follow-up is required when employing combined therapy, because the two agents balance each other’s effects. Administration of phosphate alone will lead to secondary hyperparathyroidism, and administration of 1,25(OH)2D alone commonly leads to hypercalciuria and hypercalcemia. Hence a patient on combined therapy can develop hypercalciuria as a consequence of either too much 1,25(OH)2D or too little phosphate. Because of this balance, any time it becomes necessary to interrupt phosphate administration, it is necessary to discontinue 1,25(OH)2D therapy as well.
Diarrhea and gastrointestinal upset frequently develop from phosphate administration. To minimize diarrhea, the phosphate dose should be increased gradually. If diarrhea develops, it is best to decrease (or stop) the dose of phosphate (and calcitriol; see earlier) and slowly increase therapy as tolerated. Since hypercalcemia may develop from calcitriol therapy, patients should be given instructions on the symptoms of hypercalcemia, such as depression, confusion, anorexia, nausea, vomiting, polyuria, and dehydration. Serum biochemistries should be measured if a patient experiences any symptoms of hypercalcemia. It should not be assumed that patients on high-dose calcitriol who develop nausea, vomiting, and lethargy have food poisoning or another benign etiology of their symptoms until proven otherwise.
The most common and potentially serious complication of therapy is the development of nephrocalcinosis.142–147 With the widespread use of renal ultrasound, nephrocalcinosis has been noted with increasing frequency. In most cases, the nephrocalcinosis is observed without evident changes in glomerular filtration rate (GFR). Its occurrence may be related to phosphate dosage, and some authors assert that phosphate administration leads to hyperabsorption of oxalate, which results in nephrocalcinosis.148 However, this assertion remains unproven, and patients who received vitamin D2 but no phosphate can develop nephrocalcinosis.149 Thus hypercalciuria and hypercalcemia may contribute to (or at least aggravate) this problem. Kidney biopsy specimens from three treated patients with nephrocalcinosis showed that renal calcifications are located mainly intratubularly and are composed exclusively of calcium phosphate.150 Of note, a report of two patients with nephrocalcinosis for 2 decades indicates that it was not associated with impaired renal function.149 Although these data are somewhat reassuring, the available data are not strong enough to completely allay concerns about renal complications of even carefully monitored therapy, and we are aware of at least one XLH patient with end-stage renal failure after long-term therapy.
A second, less common complication is the development of tertiary hyperparathyroidism.151–154 Presumably, tertiary hyperparathyroidism results from chronic stimulation of the parathyroid glands by phosphate therapy. Although this complication is more likely in patients who are getting too much phosphate, it also can occur in optimally treated individuals. In tertiary hyperparathyroidism, renal function may decline, necessitating a reduction in the dose of phosphate and calcitriol or cessation of therapy. This will lead to the redevelopment or worsening of the bone disease. A small number of such patients have undergone total parathyroidectomy with autotransplant to the forearm. In a number of these patients, the transplanted tissue has shown a propensity to proliferate and lead to the redevelopment of tertiary hyperparathyroidism. Removal of this hyperplastic tissue from the forearm site leads to a reduction in serum PTH levels. Theoretically, total parathyroidectomy followed by treatment with 1,25(OH)2D and phosphate is a potential option. However, this approach is not recommended, because it results in lifelong hypoparathyroidism.
Although treatment complications cannot be prevented entirely, careful monitoring may minimize treatment-related problems. Serum calcium, phosphorus, and creatinine levels and urine calcium and creatinine concentrations should be monitored monthly while patients are on high-dose therapy and every 3 to 4 months while on maintenance therapy. A 24-hour urine phosphorus measurement is often useful to gauge compliance with therapy. Serum PTH concentration should be measured at yearly intervals as appropriate. We have not seen patients with elevated urinary oxalate caused by therapy. A renal ultrasound should be obtained as a baseline at the start of therapy and periodically thereafter.
In light of the difficulty with combined therapy and the complications attributable at least in part to therapy, additional therapeutic options are being explored. These include the use of human GH,155 24,25(OH)2D,156 and diuretics.157 None of these are alternatives to combined therapy, but each has been employed as adjuvant therapy.
A continued search for better therapeutic agents is needed because it is increasingly clear that the present-day combined therapy is not curative and is accompanied by a high complication rate. However, if therapy with combined phosphate and 1,25(OH)2D is monitored carefully, growth rates increase, rickets is corrected, and bowing of the lower extremities is significantly reduced.137,139,147,158–161
Autosomal Recessive Hypophosphatemic Rickets (OMIM No. 241520)
Dentin matrix protein-1 (DMP1) is a member of the small integrin-binding ligand, N-linked glycoprotein (SIBLING) family and is highly and specifically expressed in osteocytes. Both Dmp1-null mice and patients with ARHR manifest rickets and osteomalacia with isolated renal Pi wasting associated with elevated FGF23. Mutational analyses revealed that one ARHR family carried a mutation that ablated the DMP1 start codon (Met1Val), and a second family exhibited a deletion in the DMP1 C-terminus (1484-1490del).162 Mutations have also been identified in DMP1 splicing sites, which likely result in nonfunctional protein.163
Mechanistic studies using the Dmp1-null mouse have demonstrated that loss of Dmp1 impairs osteocyte maturation, leading to markedly elevated FGF23 mRNA and protein expression. The hypophosphatemia results in pathological changes in bone mineralization,162 which can be largely but not completely abrogated by high-phosphate diet in Dmp1-null mice. Importantly, Dmp1-null mice are biochemical phenocopies of the Hyp mouse, and patients with ARHR and XLH (as well as the Dmp1-null and Hyp mice) share a distinctive bone histology characterized by periosteocytic lesions of nonmineralized bone.162 Thus, these findings suggest that PHEX (mutated in XLH) may also have a role in osteocyte maturation in a parallel pathway to DMP1 that leads to overexpression of FGF23.
Other Disorders Involving FGF23
Fibrous Dysplasia
Fibrous dysplasia (FD) is a disorder caused by activating somatic mutations in the GNAS1 gene, encoding the α subunit of the stimulatory G protein, Gs.164,165 The skeletal defects in FD are characterized by fibrous lesions and co-localized mineralization defects, which contribute to the morbidity in these patients.166 A significant proportion of FD patients also manifest various degrees of renal Pi wasting and subsequent hypophosphatemia,167 which can lead to hypophosphatemic rickets and osteomalacia. Extraskeletal clinical manifestations of FD can occur, such as abnormal skin pigmentation, premature puberty, and hyperthyroidism; the disease is then referred to as McCune-Albright syndrome (MAS). In one study of FD patients, increased FGF23 serum levels correlated negatively to serum Pi and 1,25(OH)2D3 but positively to skeletal disease burden.109 FGF23 mRNA and protein were localized to fibrous cells in the fibrous bone lesions of FD, as well as osteogenic and endothelial cells associated with microvascular walls in bone.109 Therefore, it is likely that FGF23 plays an important role in the pathogenesis of the phosphate wasting that is often seen in FD/MAS.
FD patients are often treated with bisphosphonates, which can lead to improvement of symptoms. It was reported that several MAS patients significantly reduced their elevated FGF23 levels after pamidronate therapy, further supporting a central role of FGF23 in FD/MAS.168 The mechanisms of action for this reduction of serum FGF23 levels is unclear, but one plausible explanation is that osteogenic cells overproducing FGF23 in FD/MAS may undergo apoptosis, thus leading to decreased production of FGF23.
The reason why the bone cells associated with lesions in FD overexpress FGF23 is not understood. Normally, FGF23 is expressed in osteogenic cells,109,110 but it is possible that improperly differentiated cells of FD lesions may have lost regulatory mechanisms required to suppress FGF23 production. Speculatively, this is analogous to FGF23 overexpression in TIO tumors, which are not subject to normal regulatory mechanisms.169
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


