FIGURE 66-1. Schematic representation of the main steps of the vitamin D biosynthetic pathway, where genetic aberrations may lead to rickets and osteomalacia. The renal defect in pseudo–vitamin D–deficiency rickets (PDDR) is indicated by the break in the 1,25(OH)2D3 arrow arising in the kidney. The mutation leads to insufficient synthesis of 1,25(OH)2D. The left part of the figure represents a target cell where schematic coupling of the ligand to its receptor (VDR) takes place in the cytosol or, more likely, in the nucleus. The VDR then heterodimerizes with the RXR receptor. For ease of representation, the RXR ligand (9-cis retinoic acid) is not depicted. The complex then binds to DNA to regulate gene transcription. Various mutations affecting either of the two VDR domains (DBD, DNA-binding domain; LBD, ligand-binding domain), depicted by the stippled X over the receptor complex, cause hereditary vitamin D–resistant rickets (HVDRR).
The prime metabolic consequence of vitamin D deficiency is reduced net intestinal absorption of calcium.1,2 Calcium malabsorption leads to a fall in plasma calcium, secondary hyperparathyroidism, reduced renal tubular reabsorption of phosphate, hypophosphatemia, and thus a reduction in the calcium X phosphate product. Eventually, deposition of mineral in osteoid is impaired because the supply of the relevant ions is reduced. The impaired mineralization triggers the development of the rachitic and/or osteomalacic phenotype.
CLINICAL FEATURES OF PSEUDO–VITAMIN D–DEFICIENCY RICKETS (VITAMIN D–DEPENDENT RICKETS TYPE I)
The clinical symptoms of pseudo–vitamin D–deficiency rickets (PDDR), also referred to as vitamin D–dependent rickets type I, are similar to those of common vitamin D–deficiency rickets, including failure to thrive, hypotonia, and growth retardation. Affected babies lie supine because of severe muscle weakness and bone pain. At this age, gross skeletal deformities are rare; however, if diagnosis and treatment are delayed, severe deformities of the spine and long bones occur, together with generalized muscle weakness simulating myopathy. Motor problems translate into regression in head control and ability to stand. In some patients, the initial event is generalized convulsions, tremulations and Bravais-Jacksonian fits, or tetany. Pathologic fractures may occur (Fig. 66-2). The onset in most cases occurs early during the third trimester of life; the patients look healthy at birth.
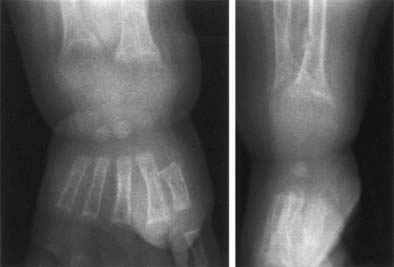
FIGURE 66-2. Radiograph of the wrist of a 19-month-old boy with pseudo–vitamin D–deficiency rickets (PDDR) at diagnosis. Severe rickets and demineralization are evident. Pronounced hypocalcemia and secondary hyperparathyroidism were documented, the latter causing a metaphyseal pseudofracture clearly seen on the lateral view.
Physical examination reveals a small, hypotonic child with wide anterior fontanel, frontal bossing, and frequent craniotabes (easy depression of the softened parieto-occipital region). Tooth eruption is delayed, and erupted teeth show evidence of enamel hypoplasia. A rachitic rosary is either visible or palpable. In limbs, widening of the metaphyseal areas is evidenced by enlargement of wrists and ankles, and there is a variable degree of deformity (bowing) of long-bone diaphyses. Deformities of the thorax may interfere with ventilation and predispose to pulmonary infection; infant death by pulmonary infections was not infrequent in the past, when the diagnosis was either missed (confused with a neurologic or respiratory condition) or made too late. The Chvostek sign (twitching of the upper lip upon light finger tapping of the facial nerve) reflects nerve irritability, a consequence of a rapid fall in serum calcium.
X-ray features include diffuse osteopenia (mild to severe hypomineralization of the skeleton) and classic rachitic metaphyseal changes: fraying, cupping, widening, and fuzziness of the zone of provisional calcification immediately under the growth plate (see Fig. 66-2). These changes are seen better and detected earlier in the most active growth plates—namely, the distal ulna and femur and the proximal and distal tibia. Changes in the diaphyses may not be evident when metaphyseal changes are first detected. However, they will appear a few weeks later as rarefaction, coarse trabeculation, cortical thinning, and subperiosteal erosion. Looser-Milkman’s pseudofractures and curvature of the shafts of long bones may be observed, especially in children older than 1 to 2 years.
Hypocalcemia is the main biochemical feature in PDDR. A rapid decrease in serum calcium concentration may give rise to tetany and convulsions, which may occur prior to any radiologic evidence of rickets. Prolonged hypocalcemia triggers secondary hyperparathyroidism and hyperaminoaciduria.21 Urinary calcium excretion is very low, whereas fecal calcium is high as a consequence of impaired intestinal calcium absorption. Elevated urinary cyclic adenosine monophosphate (cAMP) is not a consistent finding, and normal values have been measured in PDDR patients, despite high circulating parathyroid hormone (PTH) levels.22
Serum phosphate concentrations may be normal or low. When reduced, the hypophosphatemia is usually of a lesser degree than that measured in X-linked hypophosphatemic rickets.23 It results from the combination of impaired intestinal absorption and increased urinary loss induced by the secondary hyperparathyroidism. Serum alkaline phosphatase activity is consistently elevated, and its increase precedes the appearance of clinical symptoms.
Patients with PDDR have normal serum levels of 25(OH)D after exposure to sunlight or oral intake of small doses of vitamin D; the concentrations increase if higher doses are given.22 Circulating levels of 24,25-dihydroxyvitamin D [24,25(OH)2D] are normal and correlate with those of 25(OH)D.24 Serum levels of 1,25(OH)2D are low in untreated patients.22,25 This is evident immediately after birth, months before any clinical evidence of rickets appears. Even when patients are treated with high doses of vitamin D, causing major increases in circulating levels of 25(OH)D, the blood concentration of 1,25(OH)2D does not reach the normal range. These characteristic features of serum vitamin D metabolites have provided key insight into the pathogenesis of PDDR.
CLINICAL FEATURES OF HEREDITARY VITAMIN D–RESISTANT RICKETS (VITAMIN D–DEPENDENT RICKETS TYPE II)
Many of the clinical findings in patients with hereditary vitamin D–resistant rickets (HVDRR), also termed vitamin D–dependent rickets type II, are identical to those described for PDDR, including bone pain, muscle weakness, hypotonia, and occasional convulsions.26 Children are often growth retarded, and hypoplasia of the teeth is observed. The radiologic features of rickets are present. A major difference is that many children with HVDRR have sparse body hair, and some have total scalp and body alopecia, sometimes even including eyebrows and eyelashes. Hair loss may be evident at birth or occur during the first months of life. Patients with alopecia generally have more severe resistance to vitamin D. In families with a prior history of the disease, the absence of scalp hair in newborns can provide initial diagnostic clues for HVDRR. A defect in epithelial-mesenchymal communication that is required for normal hair cycling has been shown to be the cause of the alopecia in an animal model of HVDRR.27
Serum biochemistry includes low concentrations of calcium and phosphate and elevated alkaline phosphatase activity. Secondary hyperparathyroidism with elevated circulating PTH is measurable. The key difference concerns circulating levels of vitamin D metabolites. The 25(OH)D values are normal, and in the cases in which it has been measured, 24,25(OH)2D levels have been low. Importantly, serum levels of 1,25(OH)2D are elevated. This clinical feature clearly distinguishes HVDRR from PDDR, where circulating concentrations of 1,25(OH)2D are depressed. Additionally, patients with HVDRR are resistant to supraphysiologic doses of all forms of vitamin D therapy. Table 66-1 outlines the similarities and differences between the two forms of hereditary rickets involving the vitamin D endocrine system.
Table 66-1. Comparison of Pseudo–Vitamin D–Deficiency Rickets (Vitamin D–Dependent Rickets Type I) and Hereditary Vitamin D–Resistant Rickets (Vitamin D–Dependent Rickets Type II)
| Feature | PDDR (or VDDR-I) | HVDRR (or VDDR-II) |
|---|---|---|
| Mutations | CYP27B1 (25[OH]D-1α-hydroxylase) | VDR (vitamin D receptor) |
| Genetic inheritance | Autosomal recessive | Autosomal recessive |
| Age of onset | Early | Early |
| Rickets | Yes | Yes |
| Hypocalcemia | Yes | Yes |
| Serum alkaline phosphatase | Elevated | Elevated |
| Secondary hyperparathyroidism | Yes | Yes |
| Alopecia | No | Yes |
| Serum 25(OH)D | Normal | Normal |
| Serum 1,25(OH)2D | Low | Elevated |
| Response to 1,25(OH)2D therapy | Yes | No |
Reference range, serum biochemistry (child): calcemia (total calcium), 2.2–2.7 mmol/L; alkaline phosphatase, 20–150 U/L; 25(OH)D, 35–200 nmol/L; 1,25(OH)2D, 12–46 µmol/L. HVDRR, Hereditary vitamin D–resistant rickets; PDDR, pseudo–vitamin D–deficiency rickets; VDDR-I, vitamin D–dependent rickets type I; VDDR-II, vitamin D–dependent rickets type II. From Favus MJ: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3rd ed. Philadelphia: Lippincott-Raven, 1996, pp 451–452.
Pseudo–Vitamin D–Deficiency Rickets
MOLECULAR ETIOLOGY
As previously mentioned, serum levels of 25(OH)D are normal in untreated patients with PDDR and elevated in patients receiving large daily amounts of vitamin D.22 These results indicate that intestinal absorption of vitamin D and its hydroxylation in the liver are not impaired in PDDR. Circulating levels of 24,25(OH)2D are also normal and highly correlated with those of 25(OH)D, indicating a fully functional 25(OH)D-24-hydroxylase enzyme.24 However, serum concentrations of 1,25(OH)2D are low in untreated patients and remain low even when they are treated with high doses of vitamin D.22,25 This clearly identifies defective activity of the 25(OH)D-1α-hydroxylase enzyme (CYP27B1; hereafter referred to as 1α-hydroxylase) as the basic abnormality in PDDR and differentiates it from HVDRR.
PDDR is inherited as a simple autosomal recessive trait.28 No phenotypic abnormalities are observed in heterozygotes.21 By taking advantage of the unusual frequency of PDDR in the French-Canadian population and the availability of sample material from relatively large kindreds, the PDDR locus was mapped to the region of band 14 on the long arm of chromosome 12 (12q13-14).29,30
Tremendous progress has been achieved in the study of the molecular etiology of PDDR through the cloning of the complementary DNA (cDNA) encoding for 1α-hydroxylase.31–34 The human gene has also been cloned, sequenced, and mapped to 12q13.1-13.3 by fluorescence in situ hybridization,33,35,36 consistent with the earlier mapping of the disease to this locus by linkage analysis.
The ultimate proof that mutations in the 1α-hydroxylase gene were responsible for the PDDR phenotype required the identification of such mutations in PDDR patients and carriers of the disease. The first identified mutation was reported by Fu et al.31 in 1997; several additional mutations in various ethnic groups have since been published.35–41 These findings unequivocally establish the molecular genetic basis of PDDR as inactivating mutation(s) in the 1α-hydroxylase gene (CYP27B1). Further proof was provided by developing valid animal models of the disease using targeted inactivation of the cyp27B1 gene in mice.42,43
25-HYDROXYVITAMIN D 1α-HYDROXYLASE
Characteristics
The 25(OH)D-1α-hydroxylase (CYP27B1; 1α-hydroxylase) enzyme catalyzes the addition of a hydroxyl group at position 1α of the secosteroid backbone of 25(OH)D. The 1α-hydroxylase is a mitochondrial cytochrome P450 enzyme that requires electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to promote catalysis. These are delivered to the P450 moiety by the flavoprotein NADPH-ferredoxin reductase44 and the nonheme iron protein, ferredoxin.45 The expression of these cofactors is ubiquitous, and their genes were mapped to chromosomes 17 and 11,44,45 respectively, excluding them rapidly in the search for the PDDR mutations, since the PDDR locus was mapped early on to chromosome 12.30
The main site for the 1α-hydroxylation of 25(OH)D is the proximal tubule of the renal cortex.46 In the kidney, the expression of the 1α-hydroxylase gene is subject to complex regulation by PTH, calcitonin, calcium, phosphorus, and 1,25(OH)2D itself.47–49 The 1α-hydroxylase gene exists in a single copy in the human genome and contains nine exons spanning 5 kb of sequence. The ferredoxin-binding domain is encoded by sequences contained in exons 6 and 7, while the heme-binding domain is contained in exon 8.50,51
Mutations
To date, 35 different 1α-hydroxylase mutations have been described in PDDR patients and their parents (Table 66-2). All patients have mutations on both alleles, but a high frequency of compound heterozygosity (a different mutation on each allele) has been observed (23 compound heterozygotes out of 54 cases reported). Splice-site mutations, nucleotide deletions and duplications, and missense and nonsense mutations have been reported (see Table 66-2). The mutations are dispersed throughout the 1α-hydroxylase sequence, affecting all exons (Fig. 66-3).
Table 66-2. CYP27B1 (1α-Hydroxylase) Gene Mutations in Patients with Pseudo–Vitamin D–Deficiency Rickets (Vitamin D–Dependent Rickets Type I)
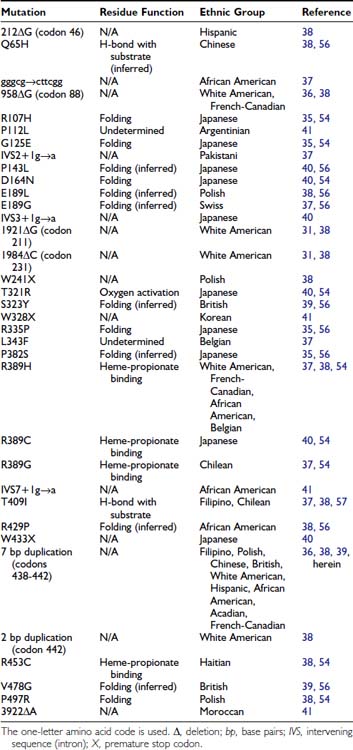
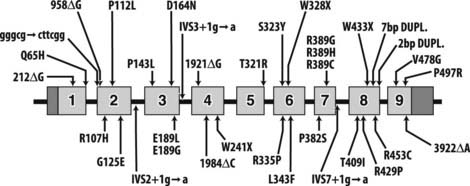
FIGURE 66-3. Mutations in pseudo–vitamin D–deficiency rickets (PDDR) patients. A schematic representation of the CYP27B1 (25[OH]D-1α-hydroxylase) gene is shown. Exons are numbered from 1 to 9 with darker shaded regions representing 5′- and 3′-nontranslated regions. Mutations are presented above and below the gene map. Numbers refer to amino acid residue; the one-letter amino acid code is used. Δ, Deletion; gggcg→cttcg, deletion of gggcg and substitution of cttcgg beginning at nucleotide 897 in exon 2; IVS2, IVS3, or IVS7+1 g→a, splice-site mutation in intron (intervening sequence) 2, 3, or 7; 7bp dupl, 7-base-pairs duplication; 2 bp dupl, 2-base-pairs duplication.
The mutations detected at the highest frequency are 958ΔG, common among French-Canadian patients (owing to a founder effect),29,36,38 and a mutation located at codons 438 to 442 in exon 8. These codons are composed of the duplicated 7-bp sequence 5′-CCCACCC CCCACCC-3′. In 11 families described to date,36,38,39,41 three rather than two copies of the 7-bp sequence are present, which alters the downstream reading frame (Fig. 66-4). Careful analysis of the correlation between ethnic origin, microsatellite haplotyping, and the presence of the 7-bp duplication mutation suggested that the mutation has arisen by several independent de novo events.36,38
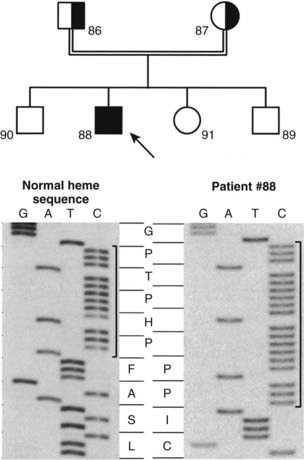
FIGURE 66-4. Identification of the molecular defect in a pseudo–vitamin D–deficiency rickets (PDDR) pedigree. Upper panel, The heterozygote parents are identified by half-filled boxes (male parent, square symbol, no. 86; female parent, round symbol, no. 87). The affected patient (male, no. 88) is identified by a filled square symbol with an arrow. Lower panel, DNA sequence analysis of the mutation within the heme-binding domain of the 25(OH)D-1α-hydroxylase (CYP27B1) gene. The 7-bp duplication is bracketed. The amino acid sequence (one-letter code) is highlighted in the center of the figure. Note the change of reading frame leading to an aberrant protein sequence and premature termination in the affected patient.
Structure/Function Relationships
An important aspect of the identification of mutations in the 1α-hydroxylase gene was to correlate the genotype of the patients with their phenotype—that is, the severity of the disease and the circulating levels of 1,25(OH)2D. In several cases, although 1,25(OH)2D serum levels are low, they are not undetectable,22,25,37,40,52 suggesting some degree of residual 1α-hydroxylase activity. Presumably, some 1α-hydroxylase mutations affect the structural integrity of the enzyme, resulting in a modification of its kinetics. This reasoning cannot apply to the frameshift (deletions, insertions, and duplications) and nonsense mutations described to date. All such mutations eliminate the heme-binding site of the protein and thus completely abolish the 1α-hydroxylase enzymatic activity. The apparent residual 1α-hydroxylase activity observed in some patients could be attributable to missense mutations. Most of these missense mutations were entirely devoid of enzymatic activity in the assays used,38,40 except for the L343F mutation (retained 2.3% of wild-type activity) and the E189G mutation (retained 22% of wild-type activity).37 Thus some missense mutations contribute to the variable phenotype observed in patients with PDDR.
Early modeling efforts16,38 compared the sequence of the 1α-hydroxylase protein (a mitochondrial class I cytochrome P450) with the sequence of bacterial class I cytochrome P450s for which x-ray crystallographic data were available. The tertiary structures of these enzymes show remarkable conservation despite low amino acid sequence identity.53 These sequence alignments yielded improper predictions of the functions of the residues mutated in PDDR patients.54 Further modeling based on the first solved crystal structure of a eukaryotic cytochrome P450,55 combined with extensive structure/function analysis of recombinant 1α-hydroxylase proteins, identified the functions of many residues mutated in PDDR patients (see Table 66-2).54,56,57 Most mutations affect folding and conformation.54,56 Residue T321 is involved in oxygen activation, and amino acid R389 is essential for heme binding.54,56 Spectroscopic analysis of wild-type and mutant 1α-hydroxylase proteins identified residue T409 as critical for binding of the 25(OH)D substrate.57
TREATMENT
Vitamin D2, at high doses, was initially used to treat PDDR. Under such treatment, circulating levels of 25(OH)D increase sharply, and it is likely that massive concentrations of 25(OH)D can bind to the VDR and induce the response of the target organs to normalize calcium homeostasis. The risk of overdose is high because vitamin D progressively accumulates in fat and muscle, and the therapeutic doses are close to the toxic doses, ultimately placing the patient at risk for nephrocalcinosis and impaired renal function. The use of 25(OH)D3 as a therapeutic agent in PDDR has been reported.58 The mechanism of action is likely to be similar to the one described earlier for vitamin D. The low availability and high cost of the metabolite have not encouraged its widespread use as long-term therapy for PDDR.
The treatment of choice is long-term (lifelong) replacement therapy with 1,25(OH)2D3.22,59 This results in rapid and complete correction of the abnormal phenotype, eliminating hypocalcemia, secondary hyperparathyroidism, and radiographic evidence of rickets. Strikingly, the myopathy disappears within days after initiation of therapy. The restoration of bone mineral content is equally rapid and histologic evidence of healing of the bone structure has been reported.22 Correction of tooth enamel hypoplasia is only partial. An important aspect of treatment is to ensure adequate calcium intake during the bone-healing phase. Needs can be monitored by frequent assessment of urinary calcium excretion. It should be noted that hypercalciuria is common during treatment with 1,25(OH)2D3, particularly during the first year of administration. Its close monitoring is used to adjust the needs in 1,25(OH)2D3. The initial dose will be 1 to 2 µg/d, and the maintenance dose will vary between 0.5 and 1 µg/d. High levels of calcium excretion may amplify the pattern of calcium deposition in the kidney, so frequent renal imaging and assessment of renal function are essential during the course of treatment.
Before 1,25(OH)2D3 became available from commercial sources, several investigators used the monohydroxylated analog, 1α(OH)D3,60 which only requires liver hydroxylation at position 25 to be activated to the hormonally active metabolite. The 25-hydroxylation step is not affected by the PDDR mutation. Response to treatment with 1α(OH)D3 is rapid, with healing of rickets in 7 to 9 weeks, and this compound is still used in several countries. On a weight basis, 1α(OH)D3 is about half as potent as 1,25(OH)2D3, nullifying any possible economic advantage in favor of the monohydroxylated compound.
Hereditary Vitamin D–Resistant Rickets
MOLECULAR ETIOLOGY
Patients with HVDRR have normal 25(OH)D and low but measurable 24,25(OH)2D serum values. The circulating levels of 1,25(OH)2D are elevated from 3 to 5 times the normal values. These biochemical findings demonstrate that all the vitamin D metabolic enzymes (25-hydroxylase, 24-hydroxylase, and 1α-hydroxylase) are active in patients with HVDRR. Most patients with the disease are resistant to all forms of vitamin D therapy. This lack of response to vitamin D treatment led Albright et al.61 to introduce the concept of hormonal resistance. The molecular basis of vitamin D end-organ resistance became clearer as the mechanism of action of the hormonal form of vitamin D was elucidated.
Ligand-binding studies first established that the 1,25(OH)2D hormone, like other sex-steroid hormones studied at the time, binds to a high-affinity receptor located in the nucleus.62 It was later discovered that this receptor could bind to DNA.63 This property was utilized to purify sufficient quantities of the VDR to raise antibodies.64,65
The observation that binding sites for 1,25(OH)2D could be detected in many tissues in addition to the classical vitamin D target tissues helped to define the etiology of HVDRR. Investigators began to study the VDR from cultured fibroblasts of patients and relatives, using ligand-binding assays, radioimmunoassays, and DNA-cellulose chromatography. Other fibroblast responses measured included induction of the 24-hydroxylase enzyme activity and vitamin D–mediated growth arrest. These methodologies led to several milestone observations. In the first studies reported, [3H]-1,25(OH)2D3 binding was undetectable in fibroblasts from HVDRR patients, and high doses of 1,25(OH)2D failed to induce the 24-hydroxylase bio-marker.66 The diminished hormone binding provided a clear rationale for the end-organ resistance reported in patients. Subsequent reports continued to describe a lack of response of the patient’s cells to 1,25(OH)2D, but some patients’ fibroblasts exhibited normal [3H]-1,25(OH)2D3 binding.67 From these early reports, it was concluded that at least two classes of HVDRR patients could be recognized: “receptor-negative” patients and “receptor-positive” patients. The development of a sensitive radioimmunoassay for the VDR protein68 demonstrated that these semantic differences were incorrect. Using this assay, it was shown that fibroblasts from so-called receptor-negative patients expressed normal levels of receptor protein. Pike et al.69 hypothesized that the VDR defect in these patients’ cells was due to a structural abnormality in the ligand-binding domain preventing [3H]-1,25(OH)2D3 from binding to the receptor, not from defective synthesis of the VDR protein. The two classes of HVDRR were more adequately described by the terminology ligand-binding positive and ligand-binding negative.
A second type of VDR structural abnormality was identified in cultured cells from patients displaying the ligand-binding positive HVDRR phenotype. The VDR from these cells showed reduced affinity for heterologous DNA as measured by DNA-cellulose chromatography.70 It was suspected that the defect in these patients would likely be a point mutation in the DNA-binding domain of the VDR. Interestingly, measurements of DNA binding affinities of the VDR from parents of ligand-binding positive patients clearly identified two forms of the VDR molecule, one with normal, wild-type affinity for DNA, and the second with reduced, defective DNA binding.70 This was the first clear evidence establishing the heterozygous state of HVDRR parents. Binding and antibody-based assays had failed to reconcile genotype and phenotype of carriers in the past.
Eventually, the purified receptor was used to obtain monoclonal antibodies that led to the cloning of the VDR cDNA from various species.71–73 In turn, the VDR genomic structure was analyzed. Eight exons comprise the entire coding region, spanning approximately 50 kb of genomic DNA.74 The translation start site is contained within exon 2.
Analysis of the VDR cDNA sequence soon established that the VDR is a member of the nuclear hormone receptor superfamily, and its mechanism of action was subsequently unraveled.15 The ligand-bound receptor forms a heterodimer with the retinoid X receptor (RXR), and the dimer contacts specific sites within the regulatory domains of responsive genes. This results in positive or negative modulation of the transcription of target genes. The VDR is essential to transduce the biological effects of 1,25(OH)2D, such as promoting calcium and phosphate transport across the small intestine and maintaining calcium homeostasis. The underlying molecular basis of the hormone resistance described in patients with HVDRR is mutation in the VDR which renders the receptor nonfunctional or less functional than the wild-type VDR. Several laboratories have independently engineered valid animal models of HVDRR by inactivating the VDR gene through gene targeting.75
HVDRR follows an autosomal recessive pattern of inheritance. Parents of patients, who are heterozygous for the mutation, show no symptoms and have normal bone development. In many cases, parental consanguinity is associated with the disease. Families often have several affected children, and males and females are affected equally.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


