In the remaining 15% of cases, the disease is caused by inborn errors in the molecular steps required for the biosynthesis of thyroid hormones, and generally it is characterized by enlargement of the gland (goiter), presumably due to elevated TSH levels.5 Thyroid dyshormonogenesis shows classical Mendelian recessive inheritance (Table 92-2).
Rarely, CH of central origin is due to hypothalamic and/or pituitary diseases, with reduced production and/or effect of thyrotropin-releasing hormone (TRH) or of the thyrotropin hormone (TSH).6
HYPORESPONSIVENESS TO THYROID-STIMULATING HORMONE
Hyporesponsiveness to TSH may be due to alterations in the TSH stimulation pathway, unresponsive TSH receptor (TSHR), or mutation in the modulating proteins downstream in the signaling pathway, such as G proteins, adenylate cyclase, or the various kinases. To date, only defects in the TSHR and Gsα have been described.
Defects in the Thyroid-Stimulating Hormone Receptor
The TSHR gene maps to human chromosome 14q31 and to mouse chromosome 12; it is encoded by 10 exons producing a 1.8-kb mRNA. The TSHR belongs to the superfamily of G protein–coupled receptors. It contains an extracellular N-terminal domain with a repetitive leu-rich motif, seven transmembrane helices, three intracellular and three extracellular loops, and an intracellular C-terminal part. The TSHR is responsible for mediating TSH action on thyroid follicular cell growth, metabolism, and function, ultimately resulting in TH synthesis and secretion.
The role of the TSHR gene in CH with TSH unresponsiveness and absence of goiter was hypothesized almost 40 years ago. Useful models for studying this autosomal recessive form of CH were offered through (1) identification of hyt/hyt mice that were affected by primary hypothyroidism with elevated TSH and hypoplastic thyroid due to a loss-of-function mutation in the Tshr gene7,8 and (2) the production of Tshr−/− mice.9
TSHR mutations in humans were identified for the first time in three siblings with CH associated with high serum TSH and normal thyroid hormone.10 The siblings were compound heterozygous, carrying a different mutation in each of the two alleles. Since this report, other mutations in the TSHR gene have been identified in several patients with thyroid hypoplasia and increased TSH secretion. All the affected individuals are homozygous or compound heterozygous for loss-of-function mutations, and consistently in the familial forms, the disease is inherited as an autosomal recessive trait. This form of CH is characterized by a “small” thyroid gland in normal position. In the case of total failure of TSHR function, the patient is severely hypothyroid because the complete lack of TSH stimulation almost completely represses the metabolic activity of the thyroid gland.11 When the TSHR has a diminished affinity to its ligand, the effect may largely be compensated for by high plasma TSH concentrations. The high TSH level in these cases does not result in an exaggerated stimulation of thyroid metabolism, and goitrogenesis is not observed.
Abnormalities in the Gs Protein Subunit
Hyporesponsiveness to TSH is found in patients with pseudohypoparathyroidism type 1a (Albright’s hereditary osteodystrophy),12 a variably expressed disorder with autosomal-dominant inheritance. The cause is a defect in the Gsα subunit (gene map locus 20q13). Gsα is involved in the stimulatory pathways of TSH and TRH as well as pathways of other hormones binding to a Gsα-coupled receptor (parathyroid hormone [PTH], gonadotropin-releasing hormone [GnRH], follicle-stimulating hormone [FSH], luteinizing hormone [LH], etc.). Several mutations have been found in these cases.13,14 Patients tend to have only mild manifestations of hypothyroidism, with normal or slightly decreased plasma free thyroxine (FT4) levels and slightly elevated TSH levels. Detection of patients with pseudohypoparathyroidism type 1a by neonatal CH screening has been reported, but it is likely that most affected newborns would be missed because their blood TSH and thyroxine (T4) concentrations would not reach the cutoff levels used in the screening programs. Otherwise, the mild hypothyroidism is just a minor component of the syndrome, and early T4 supplementation therapy is unable to prevent mental and growth retardation.
Other Causes
Hyporesponsiveness to TSH may also be caused by factors other than mutations in the TSHR or G proteins. Many families express the phenotype of resistance to TSH in the absence of a TSHR defect. In many subjects, the inheritance is dominant and the genetic cause has not yet been clarified.15 Possible candidates are factors located downstream of the TSHR/G-protein/cAMP cascade or other thyroid developmental genes.
THYROID DYSGENESIS
Athyreosis
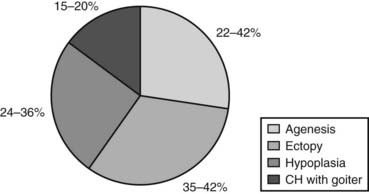
The absence of thyroid follicular cells is called athyreosis or agenesis of the thyroid. The term agenesis should be used to define the absence of the gland due to a defective initiation of thyroid morphogenesis, whereas athyreosis indicates a dysgenesis characterized by the disappearance of the thyroid following any step after the thyroid anlage specification. Athyreosis accounts for 22% to 44% of the cases of primitive permanent CH (Fig. 92-1). So far, the absence of thyroid was reported in patients with CH associated with FOXE1 gene defects (Bamforth-Lazarus syndrome),16,17 in one subject carrying a mutation in PAX818 and in one patient with NKX2-5 mutation.19
The Bamforth-Lazarus syndrome20 is a clinical entity characterized by cleft palate, bilateral choanal atresia, spiky hair, and athyreosis. Two homozygous mutations in the FOXE1 gene have been described in two pairs of siblings affected by this syndrome16,17 and in one patient with syndromic congenital hypothyroidism but not athyreosis.21 All affected members carry homozygous missense mutations in conserved amino acids within the FOXE1 forkhead domain. The mutant proteins were tested in vitro and have shown a reduction in both DNA binding and transcriptional activity.
Ectopic Thyroid
An ectopic thyroid is due to a failure in the descent of the developing thyroid from the thyroid anlage region to its definitive location in front of the trachea (see Chapter 72), therefore an ectopic thyroid can be found in any location along the path of migration from the foramen caecum to the mediastinum.
In humans, more than 50% of TD cases are associated with an ectopic thyroid (see Fig. 92-1); however, up to now, only three heterozygous mutations in the NKX2-5 gene have been linked to human ectopic thyroid.19 The functional studies of the mutant NKX2-5 demonstrated a significant functional impairment, with reduction of transactivation properties and a dominant-negative effect. The patients described were all heterozygous, and the mutations were inherited from one of the parents, suggesting that NKX2-5 mutations have variable penetrance and clinical significance.
Hypoplasia
The presence of hypoplastic thyroid has been reported in 24% to 36% of cases of CH (see Fig. 92-1). Thyroid hypoplasia is a genetically heterogeneous disease, since mutations in NKX2-1, PAX8, or TSHR genes have been reported in patients with thyroid hypoplasia.
Patients with NKX2-1 loss-of-function mutations are affected by choreoathetosis, hypothyroidism, and pulmonary alterations, with incomplete penetrance and a variability of the phenotype.22 So far, 22 loss-of-function mutations in the NKX2-1 gene have been identified in patients with this clinical picture.23–40 The unfavorable outcome in the case of impaired NKX2-1 expression, regardless of early T4 supplementation, is most likely caused by defects in the central nervous system rather than fetal hypothyroidism.
The involvement of PAX8 has been described in sporadic and familial cases of CH with TD.18,41–46 In vitro transfection assays demonstrated that the mutated proteins are unable to bind DNA and to drive transcription of the TPO promoter. All affected individuals are heterozygous for the mutations, and in the familial cases, transmission is autosomal dominant with a variable penetrance and expressivity.
Hemiagenesis
Thyroid hemiagenesis is a dysgenesis in which one thyroid lobe fails to develop. The prevalence of this morphologic abnormality ranges from 0.05% to 0.2% in healthy children, with the absence of the left lobe in almost all the cases. In these subjects, thyroid function tests are within the normal range.47
The molecular mechanisms leading to the formation of the two symmetrical thyroid lobes are still unclear, and in humans, candidate genes responsible for hemiagenesis of the thyroid have not yet been described. Indeed, Shh−/− mice embryos can display either a nonlobulated gland48 or hemiagenesis of thyroid.49 Hemiagenesis of the thyroid is also frequent in mice double heterozygous for Titf1+/− and Pax8+/−.50
DYSHORMONOGENESIS
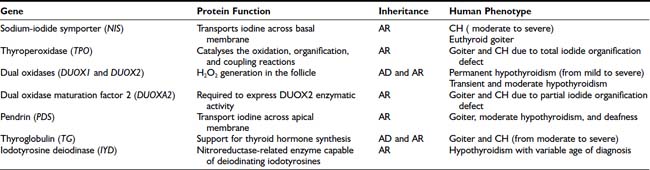
As mentioned before, in about 15% of cases, CH is due to hormonogenesis defects (see Fig. 92-1) caused by mutations in genes involved in thyroid hormone synthesis, secretion, or recycling. These cases are clinically characterized by the presence of goiter, and the molecular mechanisms in most of these forms have been identified.
In thyroid follicular cells, iodide is actively transported and concentrated by the sodium-iodide symporter present in the basolateral membrane. Subsequently it is oxidized by a hydrogen peroxide–generation system (thyroperoxidase, pendrin) and bound to tyrosine residues in thyroglobulin to form iodotyrosine (iodide organification). Some of these iodotyrosine residues (monoiodotyrosine and diiodotyrosine) are coupled to form the hormonally active iodotyronines T4 and triiodothyronine (T3), and when needed, thyroglobulin is hydrolyzed and hormones are released in the blood. A small part of the iodotyronines are hydrolyzed into the gland, and iodine is recovered by the action of specific enzymes, namely the intrathyroidal dehalogenases (see Chapter 74).
Defects in any of these steps lead to reduced circulating thyroid hormone, resulting in congenital hypothyroidism and goiter. With the exception of rare cases, all mutations in these genes appear to be inherited in autosomal-recessive fashion (see Table 92-2).
Sodium-Iodide Symporter
The sodium-iodide symporter (NIS) is a member of the sodium/solute symporter family that actively transports iodide across the membrane of the thyroid follicular cells. In 1996, NIS mRNAs from rats51 and humans52 were isolated. The human gene (SLC5A5) maps to chromosome 19p13.2-p12. It has 15 exons and encodes a 643-amino-acid protein expressed primarily in thyroid but also in salivary glands, gastric mucosa, small intestinal mucosa, lacrimal gland, nasopharynx, thymus, skin, lung tissue, choroid plexus, ciliary body, uterus, lactating mammary tissue and mammary carcinoma cells, and placenta.53,54 Only in thyroid cells iodide transport is regulated by TSH.
The inability of the thyroid gland to accumulate iodine was one of the early known causes of CH, and before the cloning of NIS, a clinical diagnosis of hereditary iodide transport defect had been made on the basis of goitrous hypothyroidism and absent thyroidal radioiodine uptake. To date, several mutations inherited in an autosomal-recessive manner have been described, with a clinical picture characterized by hypothyroidism of variable severity (from severe to fully compensated) and goiter.55,56 Thyroid morphology is heterogeneous in patients with the same NIS mutation.57
In the neonatal period, infants with iodide transport defects are found to have a normal-size or slightly enlarged thyroid gland by ultrasonography and elevated serum thyroglobulin levels.58 Radioactive iodide uptake is absent. Measurement of the saliva-to-plasma 123I ratio is around one. The degree of hypothyroidism is variable and ranges from mild to severe, possibly depending on the amount of iodide in the diet. These children are severely hypothyroid if maintained with a normal iodine diet, but addition of high amounts of iodide to the diet tends to compensate for the iodide transport failure.
Thyroperoxidase
The most frequent cause of dyshormonogenesis is thyroperoxidase (TPO) deficiency. TPO is the enzyme that catalyses the oxidation, organification, and coupling reactions (see Chapter 74).
Accumulation of iodine in the thyroid gland reaches a steady state between active influx, protein binding, and efflux, resulting in a relatively low free intracellular iodide concentration in normal conditions, but increased in the presence of TPO defects. The kinetics of iodide uptake and release can be traced by administration of radioiodide, and iodide reuptake can be inhibited by anions of similar molecular size and charge, such as perchlorate or thiocyanate. Radioiodide uptake and perchlorate inhibition give an idea of the intrathyroidal iodide concentration in relation to the circulating iodine. Iodine organification defects can be quantified as total or partial: total iodide organification defects are characterized by discharge of more than 90% of the radioiodide taken up by the gland within 1 hour after administration of sodium perchlorate, usually given 2 hours after radioiodide. A total disappearance of the thyroid image is also observed. Partial iodide organification defects are characterized by discharge of 20% to 90% of the accumulated radioiodine.59
The human TPO gene is located on chromosome 2p25 and spans approximately 150 kb; the coding sequence of 3048 bp is divided over 17 exons60 and encodes for a 933-amino-acid, membrane-bound, glycated, heme-containing protein located on the apical membranes of the thyroid follicular cell.
Defects in the TPO gene have been reported to cause congenital hypothyroidism by a total iodide organification defect, and mutations have been identified in all exons of the TPO gene. Most mutations are found in exons 8, 9, or 10, encoding the active center and heme-binding portion of the enzyme. Nonsense, splice-site, and frameshift mutations have been also described by several groups.56,61,62
If untreated, patients with organification defects show variable degrees of mental retardation, very large goiter, and hypothyroidism. In some cases with partial defects, hypothyroidism appears compensated.
DUOX1 and DUOX2
The generation of H2O2 is a crucial step in thyroid hormonogenesis. Recently two new proteins involved in the H2O2 generation in the apical membrane of the follicular thyroid cell have been identified.60 These proteins, initially named THOX1 and THOX2 (for thyroid oxidase), map to chromosome 15q15.3, only 16 kb apart from each other and in opposite transcriptional orientation. In 2001, since these proteins contain two distinct functional domains, it was suggested that they be called DUOX (for dual oxidase).
DUOX1 and DUOX2 are glycoproteins with seven putative transmembrane domains. Their function remained unclear until a factor named DUOXA2, which allows the transition of DUOX2 from the endoplasmic reticulum to the Golgi, was identified.63 The coexpression of this factor with DUOX2 in HeLa cells is able to reconstitute the H2O2 production in vitro. A similar protein (DUOXA1) is necessary for the complete maturation of the DUOX1. Interestingly, both DUOXA genes map in the 16 kb that separate the DUOX1 and DUOX2 genes on chromosome 15.
Several mutations in DUOX genes have been reported in patients with congenital hypothyroidism showing very variable phenotypes.64–67 To produce congenital permanent hypothyroidism, a severe alteration of both alleles of the DUOX2 gene is required. The presence of some residual activity in one of the alleles may produce a less severe phenotype, whereas monoallelic severe inactivation of the DUOX2 gene is associated with transient CH. In addition, the phenotype of monoallelic inactivation seems to be modulated by other factors, including environmental conditions (such as iodine insufficiency) or lifetime events (pregnancy, immediate postnatal life).
So far, no mutation in the DUOX1 gene has been identified in patients with CH. In contrast, very recently a biallelic inactivation in the dual oxidase maturation factor 2 (DUOXA2) gene has been identified in a patient with congenital hypothyroidism.68
Pendrin
In 1896, Vaughan Pendred described a syndrome characterized by congenital neurosensorial deafness and goiter.69 The disease is transmitted as an autosomal-recessive disorder. Patients have a moderately enlarged thyroid gland, are usually euthyroid, and show only a partial discharge of iodide after the administration of thiocyanate or perchlorate. The impaired hearing characteristic of the condition is not constant and is due to a cochlear defect that corresponds to the Mondini’s type of developmental abnormality of the cochlea.
In 1997, the PDS gene was cloned, and the predicted protein of 780 amino acids (86 kD) was called pendrin.70 The PDS gene maps to human chromosome 7q31, contains 21 exons, and is expressed both in the cochlea and in the thyroid. Pendrin has been localized into the apical membrane of thyroid follicular cells.71,72 In thyroid follicular cells and in transfected oocytes, pendrin is able to transport iodide.
Patients with Pendred’s syndrome are subclinically hypothyroid, with goiter, and show moderate to severe sensorineural hearing impairment. Discharge of radioiodide after administration of sodium perchlorate is moderately increased (>20%). The prevalence varies between 1 : 15,000 and 1 : 100,000.
A number of mutations in the PDS gene have been described in patients with Pendred’s syndrome.73 Despite the goiter, individuals are likely to be euthyroid and only rarely present congenital hypothyroidism. However, TSH levels are often in the upper limit of the normal range, and hypothyroidism of variable severity may eventually develop.74
Thyroglobulin
Thyroglobulin is a homodimer protein synthesized exclusively in the thyroid. The human gene is located on chromosome 8q24, and the coding sequence, containing 8307 bp,75 is divided into 42 exons.76 Following a signal peptide of 19 amino acids, the polypeptide chain is composed of 2750 amino acids containing 66 tyrosine residues. Thyroglobulin is a dimer with identical 330-kD subunits containing 10% carbohydrate residues.
Patients with disorders of thyroglobulin synthesis are moderately to severely hypothyroid. Usually, plasma thyroglobulin concentration is low, especially in relation to the TSH concentrations, and does not change after T4 treatment or injection of TSH. Patients classified in the category “thyroglobulin synthesis defects” often have abnormal iodoproteins, mainly iodinated plasma albumin, and they excrete iodopeptides of low molecular weight in the urine.77
Several mutations in the thyroglobulin gene have been reported in patients with CH78,79 and in animals, including Afrikander cattle (p.R697X),80 Dutch goats (p.Y296X),81 cog/cog mice (p.L2263P),82 and rdw rats (p.G2300R).83
Mutations in the human thyroglobulin gene are associated with congenital goiter and with moderate to severe hypothyroidism.
IYD
In addition to the active transport from the blood due to the NIS, iodine in the thyroid follicular cells derives also from the deiodination of monoiodotyrosine and diiodotyrosine.84 The gene encoding for this enzymatic activity was recently identified and named IYD (or DEHAL1).85,86 The human gene maps to chromosome 6q24-q25 and consists of 6 exons encoding a protein of 293 amino acids, with a nitroreductase-related enzyme capable of deiodinating iodotyrosines.
In the past, it was suggested that IYD mutations could be responsible for congenital hypothyroidism, but only very recently, four patients with three mutations in the IYD gene have been reported.87,88 The disease was transmitted either as an autosomal-recessive87 or an autosomal-dominant pattern of inheritance with incomplete penetration.88 Patients were hypothyroid and goitrous, with a high phenotypic variability, depending on the time of expression of the disease manifestations. The patients born after the introduction of the screening program for CH were not identified by the screening. There is also a variable severity in the clinical picture, and this can derive either from the molecular effects of the mutation (complete absence or partial activity of the protein) or from environmental factors, such as iodine diet content.
CENTRAL CONGENITAL HYPOTHYROIDISM
Central hypothyroidism is the less frequent form of CH. It occurs with an incidence of 1 in 50,000 newborns and is generally associated with alterations in hypothalamus or pituitary development.
Most patients with central CH are mildly to moderately hypothyroid. The accompanying pituitary hormonal deficiencies, especially the lack of cortisol, may be responsible for high morbidity and mortality.
Developmental Defects of the Pituitary
The pituitary gland is formed from an invagination of the floor of the third ventricle and from Rathke’s pouch, developing into the thyrotropic cell lineage and the four other neuroendocrine cell types, each defined by the hormone produced: TSH, growth hormone (GH), prolactin (PRL), gonadotropins (LH and FSH), and adrenocorticotropic hormone (ACTH).
The ontogeny of the pituitary gland depends on numerous developmental genes that guide differentiation and proliferation. These genes are highly conserved among species, suggesting crucial evolutionary roles for the associated proteins (PIT1 and PROP1, HESX1, LHX3, LHX4, and SOX3).
Lhx3 and Lhx4 belong to the LIM family of homeobox genes that are expressed early in Rathke’s pouch. In Lhx3 knockout mice, the thyrotrope, somatotrope, lactotrope, and gonadotrope cell lineages are depleted, whereas the adrenocorticotropic cell lineage fails to proliferate. This murine model shows that pituitary organ fate commitment depends on Lhx3. Lhx4 null mutants show Rathke’s pouch formation with expression of α-glycoprotein subunit, TSH-β, GH, and Pit1 transcripts, although cell numbers are reduced.
Recent studies have identified a variety of mutations in the LHX3 and LHX4 genes in patients with combined pituitary hormone deficiency diseases. These patients have complex and variable syndromes involving short stature, metabolic disorders, reproductive system deficits, and nervous system developmental abnormalities.89
Hesx1 (also called Rpx), a member of the paired-like class of homeobox genes, is one of the earliest markers of the pituitary primordium.90 Extinction of Hesx1 is important for activation of downstream genes such as Prop1, suggesting that both proteins act as opposing transcription factors.91 Targeted disruption of Hesx1 in the mouse revealed a reduction in the prospective forebrain tissue, absent optic vesicles, markedly decreased head size, and severe microphthalmia reminiscent of the syndrome of septo-optic dysplasia (SOD) in humans.
SOD is a rare heterogeneous anomaly hypoplasia of the optic nerves, various types of forebrain defects, and a variety of pituitary hormone deficiencies. Endocrine dysfunction ranges from isolated GH deficiency to complete pituitary hormonal deficiency. The human HESX1 gene maps to chromosome 3p21.1–3p21.2, and its coding region spans 1.7 kb, with a highly conserved genomic organization consisting of 4 coding exons. The first homozygous missense mutation (Arg160Cys) was found in the homeobox of HESX1 in two siblings with SOD.90 Subsequently, several other homozygous and heterozygous mutations have been shown to present with varying phenotypes characterized by pituitary hormone deficiency and SOD.91
Defects in the TRH Gene and the Thyrotropin-Releasing Hormone Receptor
In mice, homozygous deletion of the TRH gene produced a phenotype characterized by hypothyroidism and hyperglycemia.92 Only a few patients with reduced TRH production have been described in the literature,93,94 but no human mutations have been described so far.
Similarly, mice lacking the TRH receptor appear almost normal, with some growth retardation and a considerable decrease in serum T3, T4, and PRL levels but not in serum TSH.95 Thus far, only one family with a compound heterozygous96 and one family with a homozygous97 loss-of-function mutation of the TRH receptor have been described.
Defects in Thyroid-Stimulating Hormone Synthesis
TSH is a heterodimer synthesized in the pituitary gland under the control of local thyroid hormone and TRH. TSH consists of two different subunits (α and β) noncovalently linked. The TSH α-subunit is in common with LH, FSH, and chorionic gonadotropin; the β subunit is unique for TSH. The β subunit (gene map locus 1p13) synthesis is under the control of several transcription factors, including POU1F1 and PROP1. These transcription factors are the main stimulators of TSH, GH, and PRL synthesis.
Pit1/POU1F1
Pit1 (called POU1F1 in humans) is a pituitary-specific transcription factor belonging to the POU homeodomain family. The human POU1F1 maps to chromosome 3p11 and consists of 6 exons spanning 17 kb encoding for a 291-amino-acid protein. After the initial report,98 several heterozygous, compound heterozygous, and homozygous POU1F1 deletions and missense and nonsense mutations have been reported to cause this type of hereditary CH.99 Deficiency of GH, PRL, and TSH is generally severe in patients harboring mutations in POU1F1.
PROP1
Prop1 (Prophet of Pit1) is a pituitary-specific paired-like homeodomain transcription required for the expression of Pit1 and also important in regulating Hesx1 expression. Dwarf mice harboring a homozygous missense mutation in Prop1 exhibit GH, TSH, and PRL deficiency and an anterior pituitary gland reduced in size by about 50%. Additionally, these mice have reduced gonadotropin expression.100
The human PROP1 maps to chromosome 5q. The gene consists of 3 exons encoding for a 226-amino-acid protein. After the first report of mutations in PROP1 in four unrelated pedigrees with GH, TSH, PRL, LH, and FSH deficiencies,101 several distinct mutations have been identified in over 170 patients,91 suggesting that PROP1 mutations account for most cases of familial multiple pituitary hormone deficiency. Affected individuals exhibit recessive inheritance. The timing of initiation and the severity of hormonal deficiency in patients with PROP1 mutations is highly variable; diagnosis of GH deficiency preceded that of TSH deficiency in 80%. Following the deficiencies in GH and TSH, there is a delayed onset of gonadotropin insufficiency. Although most patients fail to enter puberty spontaneously, some start puberty before deficiencies in LH and FSH evolve. ACTH deficiency is a relatively late manifestation of PROP1 mutation, often evolving several decades after birth. The degree of prolactin deficiency and pituitary morphologic alterations are variable.91
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


