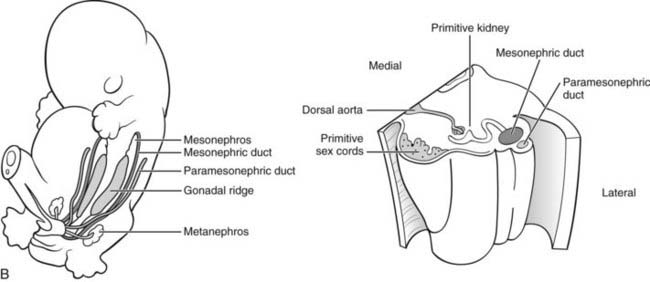
FIGURE 118-1. A, Embryology of the gonads. 1-2, Primordial germ cells are located in the wall of the yolk sac in early embryogenesis. 3, At about the fifth to sixth week of human gestation, germ cells migrate by ameboid movement from the coelomic epithelium, along the dorsal mesentery of the hindgut, to the genital ridge, the site of the future gonad. During formation of the indifferent (bipotential) gonad, the primordial germ cells infiltrate the primary sex cords, which subsequently become the seminiferous tubules of the testis or the primordial follicles of the ovary. 4-5, Formation of the testis continues after arrival of the germs cells, although these are not required for the process to occur. Probably under the direction of the newly differentiated Sertoli cells, the primitive sex cords differentiate as the seminiferous tubules, in which germ cells are embedded. Morphologic development of the testis is complete by 8 weeks’ gestation. Formation of the ovary occurs later than that of the testis, at around week 10 of human gestation, and requires the presence of germ cells. The primary sex cords develop into primordial follicles at about week 16 of gestation. B, Anatomic relationships between renal and gonadal primordia during embryogenesis. Left, Diagram of human fetus at 5 to 6 weeks’ gestation. The primordial genital ridges are located just medial to the mesonephros, the primitive fetal kidney. The mesonephros is functional only during early fetal life and is replaced later in gestation by the metanephros (located more caudally in this diagram), which becomes the definitive kidney. The mesonephric duct, which later becomes the Wolffian duct, runs in a craniocaudal direction just lateral to the mesonephros. The paramesonephric duct, which later becomes the müllerian duct, runs almost parallel to the mesonephric duct, a little farther laterally on the posterior abdominal wall. Right, Partial horizontal section of the fetus at 5 to 6 weeks’ gestation, showing (from medial to lateral) the primitive sex cords (containing primordial germ cells), primitive kidney, mesonephric duct, and paramesonephric duct.
The primitive gonadal or genital ridges initially contain no germ cells. Between the fifth and sixth weeks of gestation (mouse embryonic day [E]10.5 to 12), primordial germ cells migrate by ameboid movement from the endoderm of the yolk sac along the dorsal mesentery of the hindgut and into the bipotential gonad, where they invade the developing primary sex cords. At this stage, the gonad comprises an outer cortex and inner medulla, and there is still no morphologic difference between the gonads of male and female fetuses. Following the arrival of germ cells, the gonad begins to differentiate. In the 46,XX fetus, the cortex develops under the influence of the germ cell lineage, and the medulla regresses; in the 46,XY fetus, the reverse occurs, with development of the medullary sex cords under the influence of the supporting cell (pre-Sertoli) lineage6 (see Fig. 118-1).2 The sexually dimorphic development of germ cells is dictated in large part by retinoid signaling.7 The mesonephros produces large amounts of retinoic acid. In the developing male gonad, somatic cell expression of Cyp26b1 degrades retinoids, and the primordial germ cells undergo mitotic arrest until later in development when they resume proliferation. In contrast, the developing female gonad does not express Cyp26b1, allowing retinoids to induce a genetic program (i.e., stimulated by retinoid acid 8 [Stra8]) that results in germ cell entry into meiosis, where they arrest in meiosis I until stimulated to resume meiosis during follicle development in the adult. Female germ cells in turn play a critical role to induce somatic cell development in the ovary.
Both the urinary and genital components of the urogenital duct systems are derived to a large extent from the intermediate mesoderm, which becomes segmented into units termed nephrotomes. By about week 4 of human gestation, the lateral portions of the nephrotomes unite, forming a longitudinal duct on each side of the embryo—the mesonephric duct (later to become the Wolffian duct). At about 6 weeks’ gestation, the paramesonephric duct (forbear of the müllerian duct) forms by invagination of a tube from the epithelium on the surface of the urogenital ridge, and by 6 weeks’ gestation, both male and female fetuses are endowed with two sets of internal reproductive duct structures1,5 (see Figs. 118-1B and 118-2).
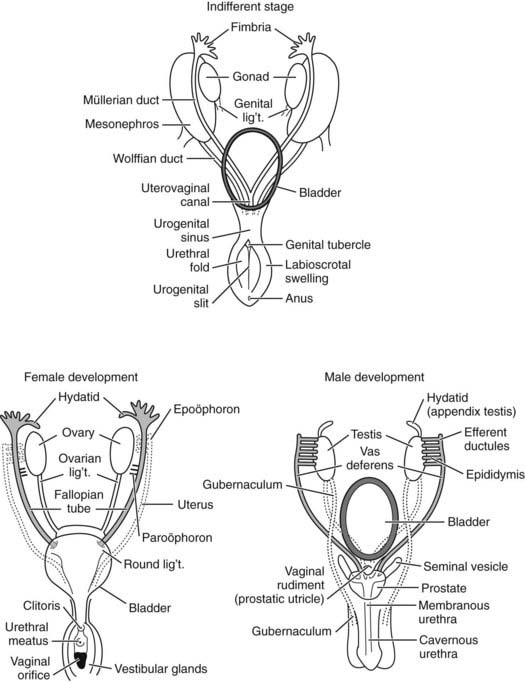
FIGURE 118-2. Embryologic development of the internal genitalia. Top, Indifferent stage. The primitive internal genitalia at 7 weeks’ gestation have both paramesonephric or müllerian (female) and mesonephric or Wolffian (male) duct systems. Lower left, Female development. In the 9- to 13-week female fetus, the Wolffian ducts regress (dotted lines), and müllerian ducts (shaded) differentiate and develop. Upper portions of müllerian ducts form fallopian tubes; lower portions fuse to form the uterus, cervix, and upper part of the vagina. The urogenital sinus remains patent in the absence of androgen action, forming the lower part of the vagina. Lower right, Male development. Differentiation of the Wolffian ducts in the male fetus occurs under the influence of testosterone between 8 and 13 weeks’ gestation. These develop as the epididymides, vasa deferentia, and seminal vesicles (shaded). By week 11, müllerian ducts (dotted lines) are obliterated by a process of apoptosis that occurs under the direction of antimüllerian hormone (AMH) secreted by Sertoli cells.
Just as the gonads and internal genitalia are indistinguishable between the sexes for the first few weeks of life, so it is for the external genital primordia. In week 4 of gestation, the external genitalia of both sexes are represented simply by a midline protuberance—the genital tubercle. By week 6, two medial folds—the urethral folds—flank the urogenital groove, and two larger folds—the labioscrotal folds—are present laterally (Fig. 118-3A).5,8 Subsequent sex-specific development is described in sections that follow.
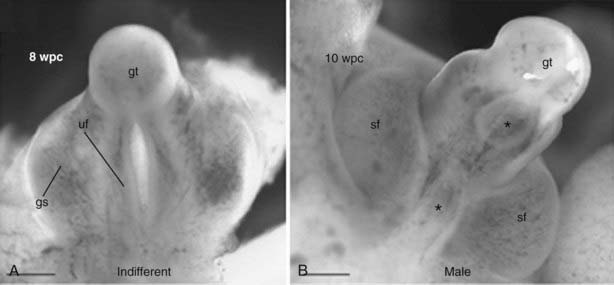
FIGURE 118-3. Embryology of external genital development. A, Indifferent stage. B, Male development. wpc, Weeks postconception; gt, genital tubercule; uf, urethral foldings; gs, genital swelling; sf, scrotal fold, *, patent regions.
(From Goto M, Piper Hanley K, Marcos J et al: In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest 116:953–960, 2006.)
GENETICS OF THE BIPOTENTIAL GONADS
A relationship between renal, adrenal, and gonadal cellular precursors underlies their development; these tissues all arise from the same regions of the primitive mesoderm and coelomic epithelium known as adrenogenital primordium.9 The steroidogenic cells of the adrenal cortex derive from mesenchymal cells attached to the coelomic cavity lining adjacent to the urogenital ridge. Similarly, the steroid-producing cells of the gonads (Leydig cells in the testis and theca cells in the ovary) differentiate from mesenchymal stem cells. Because these tissues have common cellular ancestors, it is not surprising that they share aspects of their genetic makeup. The roles of a number of transcription factors involved in the early stages of renal, adrenal, and gonadal development overlap when tissues are undifferentiated and the major developmental need is to increase cell mass. As development progresses and populations of cells begin to differentiate along specific irreversible paths, there is a requirement for a much more focused and precise program of transcription-factor action. This concept underlies the diverse roles served by transcription factors such as SF1, WT1, and LIM1, compared with the much more limited roles, expression, timing, and cellular specificity of factors such as SRY and antimüllerian hormone (AMH).
Transcription factors and other molecules implicated in the early development of the bipotential gonadal and reproductive tract primordia include LIM1, EMX2, LHX9, SF1, WT1, and WNT4. In addition, the ligand/receptor pair, Steel/c-Kit, is required, at least in mice, for the process of primordial germ cell migration from the yolk sac to the gonadal primordium in both sexes. Mouse studies have demonstrated that Emx2, Lim1, Wt1, and Wnt4 all appear to be involved in the early development of the adrenogenital primordium. Subsequent development of the specific cell lineages of the bipotential gonad involves Emx2, Lim1, Lhx9, Sf1, Wt1, Gata4, and M33. Involvement in human development is inferred on the basis of general similarities of developmental processes between species but in many cases has not been confirmed by direct evidence in humans. Exact relationships between these factors and the precise timing and order of their expression remain to be determined. A hypothetical scheme for their potential roles in regulating the processes of early development of the reproductive tract, based primarily on mouse data, is shown in Fig. 118-4B. The sections that follow provide descriptions of the molecular biology of individual factors, presented in order of currently known relevance to human development. Brief descriptions of factors with putative roles in human development, based on their known functions in mouse development, are provided at the end of this section.
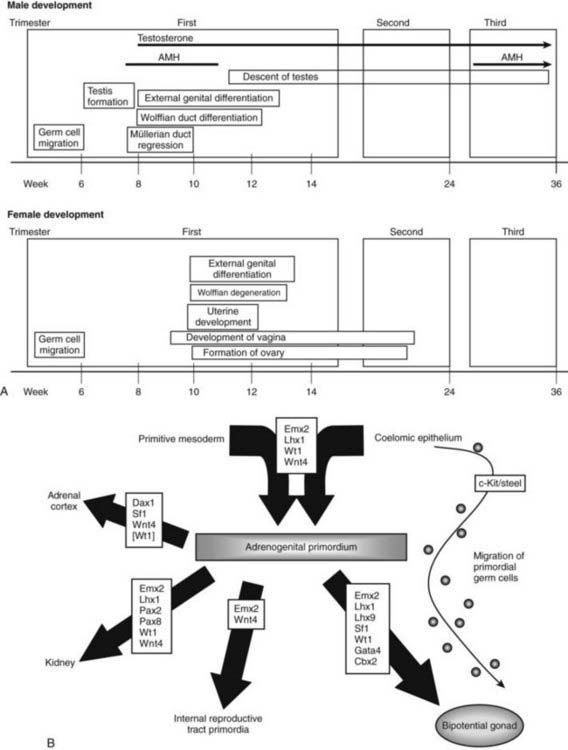
FIGURE 118-4. A, Timetable of gonadal and genital differentiation. In both male and female development, the first event of sex differentiation is the migration of germ cells at around 5 to 6 weeks’ gestation. This is followed by testicular development in the male. After onset of antimüllerian hormone (AMH) and testosterone secretion at approximately 7 to 8 weeks’ gestation, the müllerian ducts regress, and Wolffian ducts differentiate. Male development is complete by the end of the first trimester of gestation, although penile enlargement and testicular descent continue into the third trimester. The pattern of development is parallel but essentially opposite and somewhat delayed for female fetuses. In particular, there is a marked lag between testicular and ovarian development; ovarian differentiation and vaginal development continue into the middle of the second trimester. B, Genetic determinants of development of the primordial gonad. Transcription factors Emx2, Lim1, Wt1, and Wnt4 are required for development of the adrenogenital primordium, which forms following condensation of primitive mesoderm and coelomic epithelium. Thereafter, various combinations of transcription factors direct the fate of the various undifferentiated primordial cells down one of a number of pathways to form the adrenal cortex, kidney, bipotential gonad, and internal reproductive tract primordia. The roles of some of these factors are inferred from studies in mice but not yet proven in humans.
Genes With Proven Roles in Human Primordial Gonad Development
Steroidogenic Factor 1 (Nuclear Receptor Subfamily 5, Group A, Member 1—NR5A1)
The orphan nuclear receptor transcription factor, steroidogenic factor 1 (SF1; also referred to as adrenal 4–binding protein [Ad4BP] and officially termed nuclear receptor subfamily 5, group A, member 1 [NR5A1]*) appears to be one of the earliest acting and most essential factors in the primitive development of the human (and mouse) reproductive tract, affecting reproductive function at all three levels of the hypothalamic-pituitary-gonadal axis and subsequently regulating factors acting further down the pathway of gonadal/genital development in a male-specific fashion.10–12 This pleiotropic transcription factor is also critical for development of the adrenal gland. Importantly, SF1 appears to act in a dose-dependent manner in both mice and humans.13
The human SF1 gene is located at chromosome 9q33, spans 30 kb of genomic DNA, and contains 7 exons including an initial noncoding exon.14 Two distinct proteins, SF1 and ELP (embryonal long terminal repeat), are produced by alternative promoter usage and gene splicing. The key protein with respect to reproductive tract development is the 461-amino-acid, 53-kD SF1 protein iteself.15 SF1 contains two central DNA-binding zinc fingers typical of nuclear receptors, an activation domain and a carboxy-terminal ligand-binding domain proposed to bind phospholipids.16 SF1 differs structurally from most nuclear receptors by lacking an amino-terminal domain and differs functionally by binding as a monomer (rather than as the more usual dimer) to a nonpalindromic DNA steroid response element half-site, 5-AGGTCA-3′. Based on studies of human mutations, two regions of the protein appear particularly critical for function: the so-called P-box, located in the proximal portion of the first zinc finger, is responsible for interaction with the major groove of DNA; the A-box region located downstream of the DNA-binding domain modulates monomeric binding to DNA.17
SF1 is reported to regulate transcription of more than 40 genes, including those encoding the melanocortin receptor 2 (MC2R) and steroidogenic acute regulatory protein (STAR) and most genes encoding steroidogenic enzymes. Indirect evidence for such regulation includes the presence of binding sites for SF1 within the promoter regions of at least 20 target genes, including those encoding a number of other transcription factors (DAX1, WT1, SRY), hormones (luteinizing hormone [LH]β, AMH, oxytocin, Isnl3, inhibin, gonadotropin subunits), receptors (AMH-R, ACTH-R, PRL-R), proteins and enzymes of steroidogenesis (STAR, P450scc, steroid 21-hydroxylase, the aldosterone synthase isozyme of steroid 11∝-hydroxylase, aromatase), and the signaling molecule, Wnt4.17–23 SF1 up-regulates AMH expression in a sexually dimorphic manner and in doing so associates with an isoform of WT1.24–27
Expression of SF1 and DAX1 is detected in all zones of the human fetal adrenal gland as early as 4 weeks of gestation and maintained throughout gestation. Both factors play pivotal roles in adrenogonadal development. DAX1 and SF1 seem to have both antagonistic and synergistic activities.11–13 The complexity of their interaction is still not fully understood.
The gene that encodes Sf1 in the mouse is termed FtzF1, on the basis of its similarity to the Drosophila gene, fushi tarazu factor 1. Sf1 is expressed in the urogenital ridge of male and female mice at E 9 to 9.5, the earliest stage of organogenesis of the indifferent gonads.29 Consistent with its role in regulation of steroidogenesis, Sf1 is expressed prior to expression of the first enzyme of steroidogenesis, P450scc. The fact that both Sf1 and Wt1 (discussed later) are expressed in the gonadal primordium at a similar time before gonadal differentiation occurs supports the concept that Sf1 interaction with Wt1 plays a role in development of the gonadal precursor.30 After a sex-specific gonad can be discerned, Sf1 expression becomes sexually dimorphic; Sf1 expression in the Sertoli and Leydig cells of the fetal testis continues at a high level, whereas expression in fetal ovaries is quite low.29,31 Sf1 is expressed in somatic cells of XX mouse gonads until E13 and disappears altogether between embryonic days 13.5 and 16.5, in keeping with the lack of ovarian steroidogenic activity at that time. Sf1 is expressed in all primary steroidogenic tissues in adult mice, including all zones of the adrenal cortex, testicular Leydig and Sertoli cells, ovarian theca and granulosa cells, and corpus luteum.
The expression of Sf1 and Dax1, another orphan nuclear receptor described in further detail later, overlap in their tissue distribution. Dax1 appears in fact to repress or antagonize Sf1 activity, particularly the synergy between Sf1 and Wt1, probably via a protein-protein interaction with Sf1.32,33 On the basis of this finding, it has been proposed that Dax1 and Sf1 functionally oppose each other. Dax1 may function to maintain pluripotentiality, whereas Sf1 may function to induce differentiation.34
Not only does SF1 regulate gonadal and adrenal development, it also appears to control development of an important hypothalamic nucleus and pituitary gonadotropes.35 Sf1 mRNA is detectable in the developing mouse pituitary at embryonic days 13.5 to 14.5 and can be specifically detected in gonadotropes. Sf1 is also expressed in nonmammalian vertebrates including chicken and alligator, but expression patterns (and therefore likely functional roles) vary among species.
Expression of Sf1 appears to be regulated by other transcription factors, including a helix-loop-helix protein (upstream stimulatory factor)36 that binds to a region in the promoter of the gene (designated the E box) and by the homeodomain transcription factor Lhx9, which has an additive effect with Wt1 at the Sf1 promoter.36,37 Furthermore, pituitary expression of Sf1 appears to be regulated by gonadotropin-releasing hormone (GnRH), at least in the rat pituitary, and this may be relevant for the Sf1 regulation of gonadotrope development.38 However, Sf1 expression beyond the pituitary gland is not controlled by the hypothalamic-pituitary axis because hypophysectomized rats display normal Sf1 expression in the adrenals and gonads.39
SF1 mRNA is expressed widely in human tissues, both steroidogenic and nonsteroidogenic, including the adrenal cortex, gonads, spleen (primarily the reticuloendothelial cells), adult liver, heart, and brain (including several components of the limbic system).40 SF1 is expressed at high levels in the human pituitary gonadotrope and is also expressed in the human placenta from the first trimester to term.41 The diverse expression of SF1 in tissues such as spleen and brain suggests that SF1 may play a role in reticuloendothelial/immune cell maturation and/or function, as well as nervous system development and/or neurosteroid biosynthesis.
Key information regarding the developmental roles of Sf1 came initially from studies in transgenic null mice, which demonstrated the requirement for Sf1 in both sexes for development of the organs that derive from the adrenogenital primordium—primordial gonads and adrenal cortex—as well as the hypothalamus and pituitary.10,11,42 Male and female mice homozygous for deletion of Ftzf1 failed to develop steroidogenic tissues, having neither gonads nor adrenal glands. The mice had phenotypically female internal and external genitalia and died of adrenal insufficiency in the neonatal period. These mice did display mesenchymal thickening in the gonadal ridge area at the earliest stages of gonadal development (E10.5), but thereafter the cells of this region underwent apoptosis, suggesting that the role of Sf1 may be in maintaining rather than initiating gonadal development. The mice also had abnormal development of the ventromedial hypothalamic nucleus, a region important in control of pituitary gonadotropin secretion. Expression of proteins specific to gonadotrope cells—LH, follicle-stimulating hormone (FSH), and GnRH receptor—was absent, implicating Sf1 in gonadotrope development and LH/FSH expression.
The presence of female internal genitalia in the null animals of both sexes indicates that the müllerian ducts did not regress as would normally occur in the males, reflecting lack of AMH action. Because Sf1 regulates expression of AMH, deficiency of Sf1 in males would result in lack of AMH expression and therefore retention of müllerian structures. These findings imply that Sf1 is active earlier than AMH in the ontogeny of sexual differentiation. In females, expression of Sf1 during the critical period of genital development would be detrimental to normal sex differentiation, as this would lead to müllerian regression and failure of oviduct and uterine development. The fact that Sf1 expression in normal developing ovaries is low supports this contention.
In contrast to the complete absence of adrenal development in the Sf1-null mice, haploinsufficiency for Sf1 produces a different phenotype. Mice heterozygous for Sf1 deletion had significant defects in adrenal development and organization, but by compensation via cellular hypertrophy and increased expression of STAR, they were able to maintain adequate basal adrenal function.43 They developed testes and ovaries, albeit somewhat smaller than those of wild-type animals, indicating that a single active gene is adequate for this function.
Mutations identified in the human SF1 gene suggest dosage effects for the function of this transcription factor in human development. The initial descriptions of homozygous SF1 mutations in patients with 46,XY gonadal dysgenesis and female genitalia were associated with primary adrenal failure due to adrenal hypoplasia,44,45 and involved amino acid changes in the A box and P box regions of the protein. Heterozygous SF1 mutations subsequently were described in several 46,XY patients who had gonadal dysgenesis without adrenal insufficiency.46–49 It is hypothesized that because of the dose-sensitive actions of SF1, these heterozygous SF1 mutations result in a partial phenotype of 46,XY sex reversal without adrenal dysfunction. Five novel SF1 mutations were found in a cohort of 27 patients who had variable degrees of gonadal dysgenesis and severe underandrogenization.2 This finding suggests that heterozygous sequence variants in SF1 represent a relatively frequent cause of isolated 46,XY gonadal dysgenesis, with similar frequency to mutations in SRY. In contrast, heterozygous SF1 mutation in the context of a 46,XX karyotype appears to have no effect on ovarian development, as reported in a 46,XX patient with partial adrenal hypoplasia but normal ovaries, similar to the Sf1 haploinsufficiency phenotype in mice.50
Wilms Tumor 1
Like SF1, the transcription factor Wilms Tumor 1 (WT1) plays a pivotal role in early development of the primordial gonad and the bipotential internal genital duct systems prior to sex determination; one WT1 isoform appears to be required for specific development of the testis from the bipotential gonad. In renal tissue, WT1 acts as a tumor suppressor.27,51 WT1 is a member of the early growth response (EGR) family of transcription factors (proteins expressed early in the cell cycle, at G0 to G1 transition) and acts as a transcriptional activator or repressor depending on the cellular or chromosomal context.52–55 There appears to be a general requirement for WT1 in the formation of organs derived from the intermediate mesoderm, particularly the differentiation of glomerular epithelial cells and gonadal primordium.56 WT1 is implicated in gonadal and genital development by analysis of human mutations and transgenic mice with Wt1 deletions or mutations.27,60
The human WT1 gene is a complex locus at 11p13 that in fact consists of two genes, WT1 and WIT1, expressed from opposite DNA strands.58,59 The function of the WIT1 transcript is unknown, but a role as an antisense regulator of WT1 has been postulated. The highly conserved WT1 gene itself spans 50 kb and contains 10 exons that can be alternatively spliced to yield four distinct mRNA species of approximately 3 to 3.5 kb each. The primary WT1 protein is a 429-amino-acid (~50 kD) transcription factor with 4 contiguous Cys2-His2 zinc finger domains (encoded by exons 7 to 10) and an amino terminus rich in proline and glutamine, typical of certain transcription factors (Fig. 118-5A). Zinc fingers 2, 3, and 4 of WT1 have more than 60% amino acid identity with the zinc fingers of the EGR1 transcription factor. There are separate domains for transcriptional repression (AA85-124) and activation (AA181-250). These regions are distinct from the DNA-binding domain, and their activities are probably mediated by protein-protein interaction. The four mRNA species encode four major proteins, WT1 A to D, differing mainly on the basis of the presence or absence of 17 amino acids in the central region of the protein (in placental mammals) and a lysine/threonine/serine triad (abbreviated by the single-letter code for amino acids as KTS) between the third and fourth zinc fingers (see Fig. 118-5A). Perhaps as many as 32 different isoforms of WT1 result from additional variations in translational start site, either 5′ or 3′ of the main translation initiator.27,51,60 Two of the four major WT1 isoforms contain the KTS sequence (designated +KTS) and two do not (designated −KTS). The KTS amino acid triad alters the spacing between the third and fourth zinc fingers, thereby changing the DNA-binding specificity, likely preventing its binding to the typical EGR1-like DNA binding sequence. The −KTS and +KTS isoforms also have differential expression patterns within cell nuclei and appear to have distinct but somewhat overlapping roles. The fact that all transcripts are expressed at similar levels suggests that each encoded protein makes a significant contribution to WT1 function, and interactions between the proteins, each of which may have distinct targets and functions, may be important in the control of cellular proliferation and differentiation exerted by WT1. The smaller −KTS isoform has greater transcriptional activation potential than the +KTS isoform.
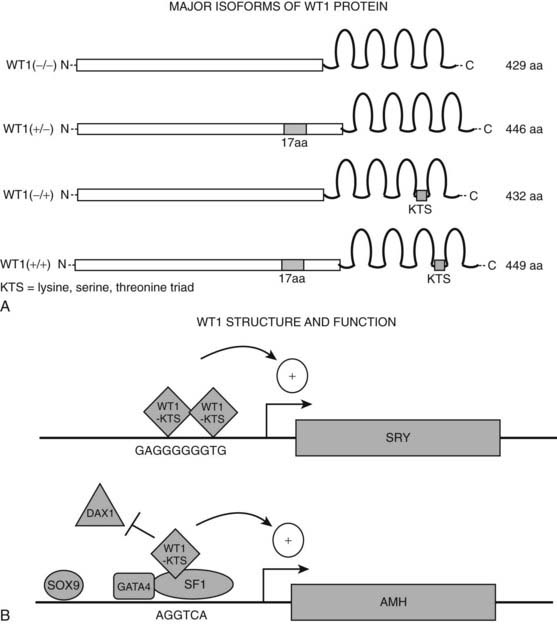
FIGURE 118-5. Structure and function of transcription factor WT1. A, Major isoforms of WT1. WT1 is a potent transcriptional activator or repressor, depending on the specific cellular and genetic context. Four major isoforms of WT1 are formed by differential splicing of exons 5 and 9. They either include or exclude a 17-amino-acid sequence in the central region of the protein (encoded by exon 5) and a lysine-serine-threonine triad (KTS) between the third and fourth zinc fingers (encoded by exon 9) that alters the DNA-binding specificity of the protein. The −/− isoform, lacking both inserts, has greater transcriptional activation potential than the main isoform (+/+) that contains both inserts. B, WT1 binding at target genes. WT1 regulates transcription of numerous target genes involved in various aspects of sex determination and differentiation, including SRY, DAX1, and AMH. WT1 (−KTS) binds as a homodimer to the promoter of the SRY gene to up-regulate its transcription. In contrast, at the AMH gene, WT1-KTS binds as a heterodimer with SF1, antagonizing binding of DAX1 to the promoter. aa, Amino acids.
Various forms of WT1 regulate SRY, DAX1, SF1, and AMH expression (Fig. 118-5B).33,61–63 WT1 −KTS isoforms associate and synergize with SF1 to promote AMH expression; WT1 −KTS can also activate the DAX1 promoter. However, DAX1 antagonizes the synergy between SF1 and WT1, most likely through a direct protein-protein interaction with SF1, suggesting that WT1 and DAX1 functionally oppose each other in testis development by modulating SF1-mediated transactivation. The fact that WT1 can up-regulate expression of DAX1, which in turn antagonizes WT1/SF1-mediated stimulation of AMH expression, suggests that the relative dosages of WT1 −KTS and DAX1 and the timing of their expression during embryogenesis are vital in the delicate balance of transcription factor activity required for gonadal development. The −KTS isoforms of WT1 also up-regulate SRY gene expression through the EGR1-like sequence in the core promoter.
Expression patterns for Wt1 in normal mouse embryogenesis support a role for Wt1 in development of urogenital tissues, as expression occurs very early at about the same time as Sf1 and before Dax1.64–66 Wt1 is faintly detectable as early as day 9 and readily detectable in pronephric and mesonephric tissues on embryonic day 10.5, at which time Sry expression (discussed in the section on testis development) is detectable in pre-Sertoli cells. By embryonic day 11.5, the nephrogenic cord, condensing metanephric tissue, and urogenital ridge display high levels of Wt1 message. In the developing gonad, Wt1 expression is localized to the developing sex cords; in mature gonads, it is confined to the Sertoli cells and tunica albuginea of the testis and the granulosa and epithelial cells of the ovary. The role for Wt1 in development of the primordial gonad is fairly clear, but its roles in specific testis or ovary development are less well understood. It seems likely that the various forms of Wt1 may function differentially in the different genetic contexts of testicular versus ovarian development. Consistent with this concept, Wt1 expression is differentially regulated during development, depending on the sexual differentiation of the gonad.
The expression pattern of WT1 in human fetal development is similar to that seen in the mouse during the period between days 28 to 70 of gestation, equivalent to mouse embryonic days 10 to 15.51,65 During this time, WT1 is expressed mainly in mesodermally derived tissues—kidneys, gonads, and mesothelium—but is also expressed in spinal cord and brain—tissues of ectodermal origin. In midtrimester human embryos, there is strong expression in kidneys and gonads. WT1 expression is limited to Sertoli cells in adult testes.
Transgenic mice homozygous for a knockout mutation of the entire Wt1 gene had failure of renal and gonadal development in the initial study, with additional failure of adrenal gland development in a later study.56,57,67 Specifically, at day 11 of gestation, the cells of the metanephric blastema underwent apoptosis, the ureteric bud failed to grow out from the Wolffian duct, and the formation of the metanephric kidney did not occur. The mice were nonviable, probably due to abnormal development of the mesothelium, heart, and lungs. A later study also demonstrated a requirement for Wt1 in the development of epicardium, adrenal gland, and throughout nephrogenesis.
The developmental phenotype of mice with Wt1 gene knockouts varies depending on the specific isoform of the transcription factor targeted63 and the magnitude of loss of expression (i.e., heterozygous versus homozygous deletion). A single wild-type allele is adequate for urogenital normal development, as demonstrated by the finding of normal urogenital tracts in mice heterozygous for a reduction of either the +KTS or −KTS isoform. In contrast, mice completely lacking the Wt1 −KTS isoform had tiny streak gonads in both males and females, associated with reduced Dax1 expression. The mice also had abnormal development of the internal genital ducts and severely impaired renal development. Male mice homozygous for deletion of the +KTS isoforms (which retain normal levels of the −KTS isoforms) showed complete XY sex reversal, their embryonic gonads having the morphologic appearance of ovaries, associated with a dramatic reduction of gonadal Sry and Sox9 expression and female-type Dax1 expression. These findings imply a key role for the +KTS isoforms in regulation of the essential testis-determining Sox genes in male gonadal development. Female mice lacking the +KTS isoforms had normal ovaries, indicating that ovarian development does not require this Wt1 isoform. However, kidney development was severely impaired in both sexes. Heterozygous mice with reduced Wt1 +KTS levels develop glomerulosclerosis after 2 to 3 months, representing a model for the human disease, Frasier syndrome, indicating that Wt1 +KTS dosage is important for normal postnatal renal function. These data demonstrate distinct functions for the Wt1 +/−KTS isoforms and place the Wt1 +KTS variants as likely regulators of Sry in the sex determination pathway.
It appears from these different transgenic experiments that some form of Wt1 is absolutely required for primordial gonad development, but the subsequent more specific development of the testis requires the +KTS isoform. The absence of both sets of internal genital duct anlagen in one patient with a WT1 mutation are also compatible with a primary role of WT1 in early development of the bipotential internal duct systems, although this finding has yet to be confirmed.67
Human mutations confirm the developmental importance of WT1. The Denys-Drash syndrome is associated with heterozygous germline mutations in WT1 (mainly in the zinc finger encoding regions) in more than 90% of cases. The disorder is genetically dominant; no patients have been described with mutations in both alleles of the gene. Complete deletion of WT1 produces milder genital abnormalities (cryptorchidism and/or hypospadias in 46,XY individuals) than does a mutation that encodes expression of an abnormal WT1 protein (46,XY sex reversal with streak gonads), suggesting a dominant-negative mechanism of action of mutant WT1 proteins, perhaps due to abnormal DNA binding.68 Defective gonadal development has also been reported in 46,XX individuals with WT1 mutations, underscoring the role of WT1 in both male and female gonadal development. Detailed description of these mutations is beyond the scope of this chapter but can be found in a recent comprehensive review.69
Genes With Roles in Mouse Primordial Gonad Development
Lim Homeobox Gene 9
LIM transcription factors constitute a large family (at least 40 members) of proteins that are key regulators of embryonic development.70 LIM proteins carry two tandem copies of the LIM domain (the acronym stands for Lin-11, Islet-1, and Mec-3 —the three original members of the family), a unique cysteine-rich zinc-binding domain (two zinc fingers per LIM domain, for a total of four zinc fingers) involved in protein-protein interactions. The LIM domain is followed downstream by a homeodomain, a highly conserved 60-amino-acid sequence containing a DNA-binding helix-turn-helix motif.71 LIM domains can physically interact with other proteins to form protein complexes that regulate transcription.
An early acting factor in gonadal development (and other developmental processes) appears to be Lhx9, a member of the LIM family. A role for Lhx9 in development of the primordial gonad was established by the finding of complete gonadal agenesis in Lhx9-null mice of both sexes,72 apparently as a result of failure of gonadal cell proliferation at around day 12 of gestation (rather than exaggerated apoptosis). As would be expected in the presence of gonadal agenesis, the mice had high FSH concentrations and no detectable testosterone or estradiol. All null mice were phenotypically female. Uteri, oviducts, and vagina were present but atrophic, indicating that Lhx9 is not required for development of the internal genital primordia and further suggesting that optimal development of these structures requires hormonal secretions from the gonads.
Lhx9 appears to be necessary for proliferation and invasion of the epithelial (somatic) cells of the genital ridge into the underlying mesenchyme and subsequent formation of sex cords—an essential step in formation of the gonads.73 Its primary role may be in regulation of expression Sf1 (a key regulator of adrenal and gonadal development, as described earlier): Lhx9 can bind directly to the Sf1 promoter and may act synergistically with an isoform of Wt1 in activating Sf1 expression.30 Such regulation may be confined to the gonad, as evidenced by the findings in Lhx9-null mice of normal adrenal Sf1 expression in the face of minimal Sf1 expression in genital ridges.72 Lhx9 is expressed at mouse day E9.5 in epithelial and subjacent mesenchymal cells of the early gonadal ridge. Expression localizes to the interstitial region of the developing gonad as morphologic differentiation occurs, then disappears as epithelial cells differentiate into Sertoli cells and begin to express Amh. In the fetal rat ovary, Lhx9 is highly expressed in epithelial cords. Similar to the down-regulation that occurs once testicular Sertoli cells differentiate, expression is down-regulated as ovarian epithelial cells differentiate into granulosa cells during the process of folliculogenesis. Thus it appears that Lhx9 expression is inversely correlated with the degree of differentiation of mesenchymal cells of the gonad.
The human 6-exon, 10-kb LHX9 gene, located at 1q31-q32, encodes a protein with over 98% amino-acid identity with mouse Lhx9.74,75 LHX9 expression is detectable in the abdominal region of human embryos of both sexes at the time of gonad formation. It is widely expressed in adult tissues, including testis, ovary, prostate, bowel, lung, heart, liver, and leukocytes. The finding of a number of different transcripts in different human tissues suggests complex, partly sex-specific, regulatory events involving transcription initiation and alternative splicing; the predominant form is a 4.7-kb transcript. Human LHX9 mutations have not yet been reported, despite a careful search in a study of 27 patients with 46,XY gonadal agenesis or dysgenesis.75
Lim Homeobox Gene 1
Lhx1 (also known as Lim1), an early member of the Lim family that appears to be a transcriptional activator, is implicated in gonadal and renal development in mice on the basis of the findings in mice homozygous for a null mutation (knockout), or for missense mutations that alter conserved amino acids required for zinc finger structure in the LIM domains. Mutant mice were severely defective: most died in utero, and the few that survived to delivery had no heads, kidneys, or gonads.76 In a subsequent study, female Lhx1-null neonates had complete absence of the derivatives of the müllerian ducts.77
The action of Lhx1 in gonad formation is likely at the level of the primordial gonad (and primitive duct structures, discussed later), because Lhx1 expression in the mouse occurs extremely early at the stage of the primitive streak, intermediate mesoderm, and nephrogenic cord.78 Lhx1 is also expressed later in development in several urogenital tissues such as mesonephros, Wolffian and müllerian ducts, ureteric buds, and definitive kidney.77 Subsequently, mouse Lhx1 expression can be detected in the central nervous system from the telencephalon through the spinal cord and in the developing excretory system, including the pronephric region, mesonephros, nephric duct, and metanephros.79
Human Lhx1 shares over 99% identity at the amino acid level with mouse Lhx1, and the LHX1 gene is located at 11p13-p12, making it quite a close neighbor to WT1.80,81 The gene comprises 5 exons and encodes a 384-amino-acid polypeptide. The 2 LIM domains are encoded by the first and second exons; the homeodomain is encoded by exons 3 and 4. Human LHX1 expression has been detected in brain, tonsil, and thymus and also in a number of leukemia cell lines but not in adult testis or ovary; human fetal expression has not yet been reported. If this gene plays a role in human gonadogenesis, it would be expected that expression is limited to a specific time window during development of the primordial human gonad. Human mutations have not been described.
Empty Spiracles 2
Another homeodomain transcription factor, empty spiracles 2 (Emx2), has similar functions to Lhx1, being essential for the development of the dorsal telencephalon and components of the developing urogenital system in mice.82 In addition to developmental defects of the brain, Emx2-null mutant mice lacked kidneys, ureters, gonads, and genital tracts; degeneration of the Wolffian duct and mesonephric tubules was abnormally accelerated, and müllerian ducts did not form. These findings imply a role for Emx2 in very early development of the renal, gonadal, and internal genital primordia, probably at the stage of the mesonephros. Notably, Emx2-null mice had reduced expression of Lhx1, and expression of Wnt4 (described later) was absent, suggesting that Emx2 may modulate expression of these factors.82
During mouse development, Emx2 is intensely expressed in the bipotential gonads and is also expressed in epithelial components of the developing urogenital system, specifically the pronephros and mesonephros, Wolffian and müllerian ducts, ureteric buds, and the early epithelial structures derived from metanephrogenic mesenchyme.83 Emx2 is also detected in brain, kidney, and uterus. Emx2 is expressed in the adult urogenital tract in an inverse temporal pattern from Hoxa10, another homeobox gene (described later), suggesting a negative regulatory relationship.84
The 3-exon human EMX2 gene maps to 10q26.1, which is incidentally the site of a putative endometrial tumor suppressor.28,85,86 The 3-kb transcript encodes a deduced 252-amino-acid protein containing a 61-amino-acid homeodomain encoded by exons 2 and 3.
Heterozygous EMX2 mutations were reported in a study of patients with schizencephaly, a severe cerebral malformation syndrome.87 Insofar as no urogenital phenotype was described, the role of this factor in human, as distinct from murine, gonadal development remains in question. Nevertheless, sex reversal is reported in association with deletions of distal chromosome 10q, the location of the human EMX2 gene, consistent with a potential role of EMX2 in the failed testicular development of these patients.
GATA4
GATA binding protein 4 (GATA4) is a member of a group of structurally related DNA-binding zinc finger transcription factors that control gene expression and differentiation in a variety of cell types.88–91 Members of this family contain one or two zinc fingers that recognize a consensus DNA sequence known as the GATA motif (GATA[A/G],92 hence the name of this class), which is an important element in the promoters of many genes. GATA family transcription factors are critical to the development of diverse tissues. GATA4, in particular, has been implicated in formation of the vertebrate heart, gut, and gonads. With respect to sex determination and differentiation, GATA4 may, like WT1, have dual roles, being involved both in early development of the primordial gonad and later in male-specific development.
The human gene for GATA4 is located at 8p23.1-p22 and encodes a 48-kD protein that has a highly conserved zinc finger domain consisting of two zinc finger motifs and two adjacent stretches of basic amino acids.93,94 The carboxy-terminal zinc finger confers the DNA-binding ability. The protein also contains a nuclear localization domain and two amino-terminal transcription activation domains.88
The genes encoding many enzymes and proteins (including STAR) required for steroidogenesis appear to be targets of GATA proteins, because they contain the consensus GATA element within their 5′ regulatory regions.91 GATA4 and SF1 may regulate AMH expression synergistically; binding sites for both factors are adjacent to each other on the AMH promoter, resulting in protein-protein interaction via the zinc finger region of GATA4.95 GATA4 also acts synergistically with WT1 on the human SRY and AMH promoters.96 The affinities of various GATA factors for specific promoters probably depend on interactions with cofactors and other more restricted transcription factors. For example, Leydig cells express factors known as Friends of GATA, or FOG (FOG1 and FOG2), which may play a role in tissue-specific effects and also appear to be necessary for normal gonadal development in both sexes.90,97
Most tissues of mesodermal origin express either GATA4, -5, or -6 at some stage.88 In the mouse, Gata4 is expressed from early stages of gonadal development (E 11.5) in both sexes in the somatic cell population (later to become the testicular Sertoli and ovarian granulosa cells) of the indifferent gonad but not in the primordial germ cells.98,99 Abundant Gata4 expression continues in Sertoli cells throughout embryonic testis development. During early ovarian development, Gata4 mRNA and protein localize to the granulosa cells. Gata4 is markedly down-regulated shortly after the histologic differentiation of the ovary on embryonic day 13.5, resulting in a sexually dimorphic pattern of greater Gata4 expression in developing testes than ovaries. Gata4 is involved in regulating AMH activity, so reduced Gata4 expression and thereby Amh expression in the latter stages of ovary development is required for normal müllerian duct development.
In the human testis, GATA4 is expressed from early fetal development to adulthood.99 GATA4 represents an early marker of Sertoli cell function, its expression peaking at 19 to 22 weeks’ gestation.99,100 GATA4 is also expressed in a number of steroidogenic tissues, and this finding, coupled with the presence of GATA consensus elements in the promoter regions of the genes encoding many steroidogenic enzymes, is consistent with a role in regulation of steroidogenesis. In Leydig cells, GATA4 is expressed during the fetal period and after puberty, coinciding with the periods of most active androgen synthesis in the testis. Other steroidogenic tissues in which GATA4 is expressed include fetal and postnatal granulosa cells, adult theca cells, and fetal adrenal cortex (human and mouse). Fetal germ cells and prepubertal spermatogonia also express GATA4; it is down-regulated in these cells after puberty. GATA4 expression is reduced or absent in germ cells and Sertoli cells in states of androgen resistance, suggesting androgen dependence of GATA4 expression.
Because the mouse Gata4 knockout is an early-embryonic lethal mutation (day 7 to 9, before the normal onset of gonadal Gata4 expression), a gonadal phenotype of the null mutation has not been described.88 However, targeted mutations that disrupt Gata4 interactions with Fog2 (or vice versa) result in significantly reduced expression of Sry, Sox9, Amh, and Dhh, as well as the genes encoding steroidogenic enzymes P450scc, 3βHSD, and P450c17 and failure of up-regulation of Sf1 and Wt1 in XY gonads, as normally occurs in the developing testis.90,96 Wnt4, which is normally down-regulated in testicular development, was overexpressed in these mice. These abnormalities resulted in abnormal gonadal development in both XX and XY fetuses.90 Human GATA4 mutations are associated with cardiac defects, but no gonadal phenotype has been described.
Chromobox Homolog 2
Chromobox homolog 2 (CBX2), which maps to 17q25.3, is the human homolog of a mouse gene originally called M33, now called Cbx2, which appears to act as a regulator of gene expression at the chromatin level.101 Both XX and XY mice homozygous for deletion of Cbx2 had retarded formation of genital ridges, and those that survived the immediate postnatal period showed male-to-female sex reversal.102 Formation of genital ridges was disturbed in both XX and XY embryos, suggesting a role for Cbx2 in primordial gonad formation.
Sex Determination (Development of Testis or Ovary)
Once development of the primordial gonadal and genital structures has occurred, the next steps diverge between the sexes. The primary event governing the path down which morphologic sex differentiation proceeds is the development of the gonad. In the mid-20th century, the elegant experiments of Alfred Jost determined that “maleness” was a state imposed upon the fetus that would otherwise develop as a “female” (at least phenotypically)105 (see also Chapter 119). These findings led to the theory that development of the ovary and female phenotype occur when the fetus is not exposed to the influences of specific maleness-determining genes. Newer information indicates that rather than being a passive default process, ovarian/female development also requires activation of specific, perhaps opposing, gene pathways. From these findings, the concept has developed that sex determination represents the primordial battle of the sexes—the dominance of one set of gonad-specific genes over another.1 The key event determining the winner of the battle appears to be whether or not the primordial germ cells that colonize the indifferent gonad enter meiosis or are prevented from doing so by the enzyme P450 26B1 acting to degrade retinoic acid.7 Testis cord development under the influence of the SOX genes during a narrow developmental window arrests the primordial germ cells in mitosis; in the absence of testis cord development, germ cells enter meiosis, and ovarian development ensues.6,106
Sex-specific development of the gonads depends on carefully coordinated enhancement of expression of certain genes, with parallel repression of others. This coordinated expression allows for the specificity of timing and level of gene expression required to facilitate differentiation, as opposed to the more general expression and action of factors involved in early phase organogenesis. In this phase of development, the undifferentiated cell lineages—supporting cells and steroidogenic cells—that colonize the primordial gonad are directed down one of two irreversible paths, towards either testis or ovary. It makes sense that genes required for sex-specific differentiation would either be encoded on the sex-specific chromosomes or would vary in dosage between the sexes. Testis development requires that the Y-encoded gene, SRY, is active and SOX9, an autosomal gene that appears to be dose-sensitive, is active. Conversely, ovary development requires that SOX9 is repressed and WNT4 is active. The long-held concept of the “dominant” and “default” gonad is derived in part from the differential timing of development of testis versus ovary, testicular development occurring in a narrow window at least 3 to 4 weeks before ovarian development (see Fig. 118-4A). It is believed that action of male-specific genes must occur at a critical time to inhibit or repress the function of the ovary-defining genes that are otherwise destined to become active. Not only does testis development begin before ovarian development, the whole process is completed more rapidly. This phenomenon of the relative timing of development of the so-called dominant and default pathways is seen in many mammalian species, suggesting that it is a primary feature of sex determination.107 These observations suggested that mammalian sex determination is sensitive to dosage at multiple steps in its pathway.
EMBRYOLOGY OF TESTIS DEVELOPMENT
The first discernible event in testis development is the appearance of primordial Sertoli cells, which differentiate from somatic cells of the coelomic epithelium at about 7 weeks of human gestation. Soon after their appearance in the gonadal ridge, Sertoli cells proliferate, aggregate around the primitive germ cells, and align into cordlike structures that subsequently become the seminiferous tubules (see Fig. 118-1A).1,108,109 The seclusion of germ cells within the tubules prevents meiosis and commits the germ cells to spermatogenic development. The prevention of meiosis may be a key event that directs gonadal development away from the ovarian pathway, as it is believed that testis cord formation and germ cell entry into meiosis are competing pathways in gonad development. This organizational process appears to be regulated by the Sertoli cells themselves. Another key event in testis development is the differentiation of peritubular myoid cells, smooth muscle–like cells that ensheath the testis cords to provide structural integrity, thought to derive from the same interstitial cell lineage as Leydig cells. These myoepithelial cells are required for the development of the testis cords, the defining event in testicular organogenesis.110 About 1 week after Sertoli cell development (around 8 weeks in human embryos), steroidogenic Leydig cells differentiate from primitive interstitial cells of mesonephric origin. This process may be controlled by paracrine influences from Sertoli cells, possibly AMH. Germ cells are not required for the processes of testis development to occur; morphologic testis development occurs in their absence.
Finally, establishment of testis-specific vasculature enables testicular secretions to be exported to the developing fetal internal and external genitalia to direct masculinization. Initially the primitive vasculature of the genital ridge is similar in XX and XY gonads until E11.5 in the mouse. Between E11.5 and E12.5, XY gonads establish a pattern of blood flow distinct from that of XX gonads, with the development of a large male-specific coelomic vessel and a network of finely branched vessels.111 Recruitment of vascular cells from the mesonephros is essential for the development of seminiferous tubules112–114 and does not occur in XX gonads.114 The process of blood vessel development in the gonads is highly sexually dimorphic. The female gonad recruits vasculature by a classic angiogenic process. However, in the developing testis, vasculature is patterned by a remodeling mechanism that begins with the breakdown of an existing mesonephric vessel, followed by the migration and the reaggregation of XY endothelial cells to form the coelomic vessel.115 The growth rate of the vasculature is similar between XX and XY gonads, but XY gonads show a distinct arterial specification during early testis development, as demonstrated by the male-specific pattern of gonadal expression of ephrin-B2, a marker of developing arteries.116
During the second trimester of human gestation, testicular volume increases exponentially, fueled primarily by doubling of Sertoli cell numbers every 2 weeks throughout this period. This rapid increase in Sertoli cells is accompanied by a parallel increase in germ cell number and a slightly less dramatic increase in Leydig cells.100
GENETICS OF TESTIS DEVELOPMENT
Overview
Male sex determination is synonymous with testis determination. The process of testis development in the karyotypic male appears to be controlled by a switchlike mechanism that involves the Y-chromosomal SRY gene and its encoded protein, a related homeobox gene, SOX9, and no doubt other genes and proteins that either regulate or are regulated by these factors.117,118 One of the earliest effects of SRY expression is the induction of somatic cell migration from the mesonephros into the XY gonad, an essential first step in preparation for development of testis cords.108,109 Subsequently, SOX9 directs the process of seminiferous tubule organization by Sertoli cells and regulates transcription of the Sertoli cell glycoprotein, AMH; AMH in turn may play a role in directing undifferentiated interstitial cells to develop as Leydig cells.119,120 Sertoli cells act as organizers, causing surrounding cells to differentiate into either Leydig or myoid cells. In addition, Sertoli cells express Cyp26b1, a retinoid-degrading enzyme, thereby acting as a barrier to retinoid action, which would otherwise induce germ cell meiosis. Instead, male germ cells undergo mitotic arrest, preparing them for later spermatogenesis; once testicular differentiation is established, other Y-encoded genes are required to maintain spermatogenesis.
A number of other molecules have been identified as having involvement in testis development, but their exact positions in this pathway, their functions, and the factors they regulate or by which they are regulated, remain to be elucidated. These include the +KTS isoform of WT1 (as described earlier, WT1 is also crucial for development of the primordial gonad); the helicase enzyme, ATRX, involved in DNA recombination, repair, and regulation of transcription; perhaps the intracellular signaling molecule, desert hedgehog (DHH); and a gene or genes located on the distal long arm of chromosome 10. Additional X-chromosomal sequences are also likely to affect testis development, perhaps negatively, as evidenced by the fact that the presence of one or more extra X chromosomes (e.g., in Klinefelter syndrome and its variants) is associated with reduced testicular size. There are many hypothetical schemes for the interactions of the ever-expanding coterie of transcription factors involved in sex determination, but no definitive model for this process currently exists. What is clear is that the system is extremely complex, involving a network of interacting transcription factors in a nonlinear web of up-regulation or activation and down-regulation or repression steps. A simplified diagram of a possible mechanism for testis development is depicted in Fig. 118-6A. Although analyses of mouse models and in vitro cell culture assays have provided key insights, it is important to note that whereas the mouse is generally a convenient model, differences between mice and humans must not be overlooked. The details of sex determination and differentiation elucidated in rodents cannot necessarily be extrapolated to human development, and the phenotypic effects of many mutations even differ among mouse strains, emphasizing the importance of genetic modifiers. These genes and factors involved in testis development are described individually in further detail in the following sections, with particular emphasis on those genes implicated in human 46,XY gonadal dysgenesis.
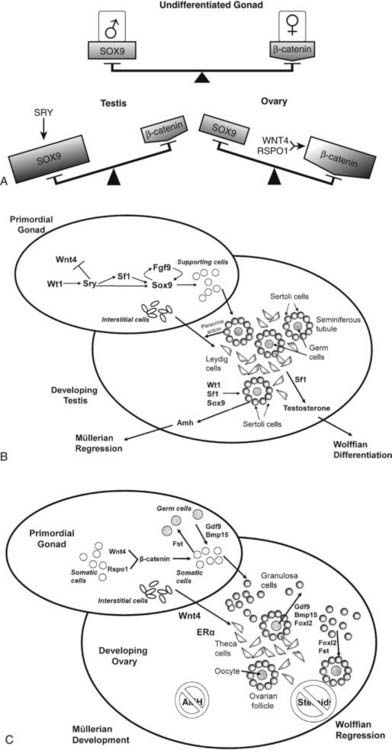
FIGURE 118-6. A, Hypothetical model of balanced signals in mammalian sex determination. In the undifferentiated gonad, the male-specific factor, SOX9, and the female-promoting molecule, β-catenin, are at similar levels of activity and balance each other out. The presence of SRY (in the XY gonad) up-regulates SOX9, thereby tipping the balance in favor of testis development by suppressing activity of the profemale factor, β-catenin. If the gonad is destined to differentiate as an ovary, WNT4 and RSPO1 activate the β-catenin signaling pathway, tipping the molecular balance in the female direction while switching off the male path. B, Hypothetical model of testis determination. The bipotential gonad of an XY fetus expresses the key testis-determining gene, Sry, which is up-regulated by Wt1. Sry in turn initiates a feed-forward loop between Fgf9 and Sox9. Fgf9 up-regulates Sox9 and is required for Sox9 maintenance, and Sox9 is required for Fgf9 up-regulation in XY gonads. A critical role of Sox9 is to stimulate differentiation of primitive supporting cells into Sertoli cells, which are the primary organizers of the seminiferous tubule. The seclusion of germ cells within the tubules prevents germ cell meiosis and commits them to spermatogenic development. Under the influence of Sertoli cells, interstitial cells differentiate as Leydig cells, subsequently secreting testosterone under the drive of Sf1. Meanwhile, Sertoli cell secretion of antimüllerian hormone (AMH) is up-regulated by the combined influences of Wt1, Sf1, and Sox9. Testis secretion of testosterone and Amh cause Wolffian duct differentiation and müllerian duct regression, respectively. C, Hypothetical model of ovary determination. In the normal XX gonad, Sry and Sox9 are not expressed. Consequently, Sertoli cells do not differentiate, and testis cords do not form. Instead, follistatin (Fst) is differentially expressed and may bind members of the transforming growth factor β (TGF-β) family, such as activins or Bmp2. Wnt4, in synergy with Rspo1, activates the β-catenin signaling pathway and blocks the male path in XX gonads. Granulosa cells surround germ cells to form ovarian follicles, perhaps under the influence of Foxl2. Meanwhile, interstitial cells are prevented from developing as Leydig cells by the action of Wnt4 secreted by supporting cells; instead, they develop as theca cells. In the absence of Leydig cells and Sertoli cells, neither testosterone nor antimüllerian hormone (AMH) is secreted. Consequently, Wolffian ducts regress, and müllerian ducts differentiate.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


