FIGURE 152-1. Communication in the gastrointestinal endocrine system. Gastrointestinal endocrine cells respond to nutrients (open-type cells) and/or stretch (closed-type cells). They may release peptides into the circulation to affect distant cells (endocrine), into the interstitial fluid to affect themselves (autocrine), or other nearby cells (paracrine) (e.g., somatostatin [SMS]). Cells in the gut may be stimulated by or influence local neurons (neurocrine effect). Neurotransmitter peptides may be released at very low levels into the circulation (spillover) and at high levels, mainly in pathologic states (e.g., vasoactive intestinal peptide [VIP] in VIPomas).
Gut peptides are single-chain polypeptides grouped together into families, based on their structures (Table 152-1). Different peptides may be derived from the same precursor peptide through tissue-specific posttranslational processing (e.g., proglucagon-derived peptides) or from the same gene through differential splicing (e.g., calcitonin gene–related peptide [CGRP]; calcitonin). Peptide precursor molecules undergo several intracellular modification steps that may include signal peptide cleavage, tyrosine sulfation, dibasic cleavage, removal of basic residues, and C-terminal amidation. Sorting of peptides into secretory granules depends on structural domains within their molecule. Most gut peptides are released in response to meals, with the degree of release depending on meal type and size. Gut peptides act through seven transmembrane G protein–coupled receptors. The specific tissue expression of these receptors, the existence of different receptor subtypes and receptor modifying proteins, and coupling to different secondary messenger mechanisms allow specificity and variability in gut peptide action. Gut peptides are usually broken down by circulating enzymes and metabolized and excreted by the liver and kidney. Fig. 152-2 shows the half-lives of various gut peptides. Circulating levels of gut peptides vary with different gastrointestinal diseases; this may be due to abnormal changes in release from the damaged bowel or adaptive compensatory changes in the remaining healthy bowel. Common causes for elevated gut peptides are listed in Table 152-2.
Table 152-1. Gastrointestinal Regulatory Peptides and Related Peptides
Secretin Family |
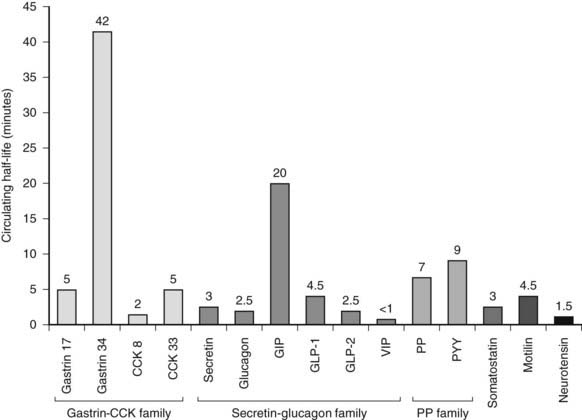
FIGURE 152-2. The circulating half-lives of gastrointestinal hormones. CCK, Cholecystokinin; GIP, gastric inhibitory peptide; GLP, glucagon-like peptide; PP, pancreatic polypeptide; PYY, peptide YY.
Table 152-2. Causes of Increases of Some Commonly Measured Gut Hormones
| Hormone | Causes of Raised Plasma Concentration |
|---|---|
| General | Postabsorptive state |
| Renal failure | |
| Hepatic failure | |
| Stress | |
| Gastrin | Drugs: H2 antagonists, proton pump inhibitors |
| Helicobacter pylori infection | |
| Pernicious anemia | |
| Hypercalcemia | |
| Intestinal resection | |
| Inflammatory bowel disease | |
| Antral remnant | |
| Gastrinoma | |
| Glucagon | Drugs: oral contraceptives |
| Diabetes mellitus | |
| Hypoglycemia | |
| Familial hyperglucagonemia | |
| Glucagonoma | |
| Pancreatic polypeptide | Inflammatory bowel disease |
| Other neuroendocrine tumors | |
| PPoma | |
| Elderly | |
| Pernicious anemia | |
| Somatostatin | Somatostatinoma |
| VIP | VIPoma |
| Hepatic cirrhosis | |
| Bowel ischem |
The gut contains a diffuse population of endocrine cells, some of which (open-type cells) communicate with and are sensitive to changes in the chemical environment of the gut lumen, such as pH and amino acids. Other endocrine cells (closed-type) lie within the gut wall and may perform other functions (paracrine, sensitive to stretch, trophic). Endocrine cells are constantly generated from stem cells. They monitor and modify digestive processes. In addition to endocrine cells, there are a wide variety of peptide-producing gut neuronal cells. Several markers are used to identify neuroendocrine cells in the gut, including chromogranins, secretogranins, neuron-specific enolase, and subtilase preprotein convertases.
This chapter will briefly describe the major gastrointestinal regulatory peptides, which have been divided into those that subserve predominantly hormonal and paracrine actions and those that act mainly as neurotransmitters. The final section deals with the rare gastropancreatic neuroendocrine tumors and their syndromes.
Physiology
HORMONES
Gastrin and Cholecystokinin
Gastrin and cholecystokinin (CCK) belong to the same family and act at the same receptors, CCK1 and CCK2.3 CCK1 (also CCK-A) is predominantly the CCK receptor, while CCK2 (also CCK-B/gastrin) has affinity for both peptides but is predominantly the gastrin receptor, since gastrin circulates at much higher (10 to 100×) concentrations than CCK.
Gastrin
Gastrin, the main hormonal stimulus to gastric acid secretion, is produced in the upper small intestine and in open-type G cells in the gastric antrum.4,5 Several molecular forms of gastrin are produced through the cleavage of the 101-amino-acid precursor, progastrin. There are three major circulating forms of gastrin, with N-termini of variable lengths: gastrin-34, gastrin-17, and gastrin-14. Other circulating but low concentration gastrins include gastrin-6, gastrin-52, and gastrin-71. Larger molecular forms, such as Gly-extended gastrin (Gly-gastrin), have been reported to have potential roles in cell proliferation in colon cancer.6,7 Full biological activity of gastrin resides in the C-terminal peptide and requires α-amidation of the C-terminal phenylalanine. Both gastrin and CCK end in the same pentapeptide, but they differ in that the tyrosine in position 7 of the C-terminus is sulfated in CCK. Approximately half of the gastrins in man are sulfated, but unlike CCK, sulfation of gastrin does not appear to affect biological activity. Gastrin-17 is the predominant gastrin in tissue extracts from the stomach and duodenum. Both gastrin-34 and gastrin-17 are biologically active, but gastrin-17 is the predominant circulating form after a meal. Plasma gastrin levels are raised in achlorhydria (pernicious anemia, atrophic gastritis), treatment with antisecretory drugs such as protein pump inhibitors (PPIs), G-cell hyperplasia, antral remnant after gastric surgery, infection with Helicobacter pylori, and in renal failure (see Table 152-2). Hypercalcemia can result in raised gastrin levels; therefore, hyperparathyroidism needs to be excluded first in multiple endocrine neoplasia type 1 (MEN1)8 patients before a gastrinoma is diagnosed.
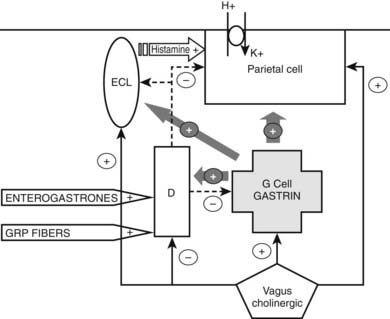
The synthesis and release of gastrin are under the control of humoral and local factors as well as the autonomic nervous system. The predominant regulatory system is the presence of amino acids, particularly aromatic amino acids, and proteins in the stomach lumen, which along with gastric distention, stimulate gastrin secretion. The vagus nerve not only stimulates gastrin secretion but also has a direct effect on acid secretion, with vagotomy resulting in increased serum gastrin and decreased acid secretion in the stomach. Postganglionic vagal fibers also release gastrin-releasing peptide (GRP), which potently stimulates gastrin secretion. The adrenergic nervous system can also influence gastrin release. Gastrin secretion is inhibited when the stomach pH falls below pH 3; this is mediated through the paracrine action of somatostatin released from neighboring antral D cells. The effect of somatostatin on G cells may be to both decrease gastrin gene transcription and increase the rate of gastrin messenger RNA (mRNA) turnover. Several peptides, including enterogastrones (factors released by the intestine that decrease gastric acid secretion and motility), act through somatostatin to reduce gastrin release. Proposed enterogastrones include vasoactive intestinal polypeptide (VIP), CGRP, glucagon, secretin, gastric inhibitory peptide (GIP), and CCK. Galanin inhibits gastrin secretion independently of somatostatin. Beer, wine, and coffee stimulate gastrin secretion independently of alcohol and caffeine.
The neural and endocrine mechanisms involved in gastric acid secretion have been studied in detail (Fig. 152-3).5 Parietal cells have three stimulatory receptors: histamine (H2), acetylcholine (M3), and gastrin (CCK2) receptors. Histamine and acetylcholine directly stimulate acid secretion, but prior histamine stimulation is required for gastrin action. The major endocrine cells involved in gastric acid secretion are the enterochromaffin-like (ECL) cells of the gastric fundus, the G cells of the antrum, and D cells of the fundus and antrum. ECL cells release histamine, which stimulates acid secretion via H2 receptors. Gastrin binding to CCK2 receptors on ECL cells and can chronically stimulate ECL cell proliferation, rarely resulting in ECL hyperplasia and carcinoids following PPI treatment. Other stimulatory receptors on ECL cells have been described: β3-adrenergic, peptidergic, and cytokine receptors. Inhibitory receptors include receptors for somatostatin and peptide YY (PYY). Somatostatin inhibits acid secretion by acting at several sites: parietal cells, G cells, and ECL cells. Fundal D cells are in turn stimulated by gastrin and acetylcholine. Gastrin mRNA, resulting in the production of sulfated gastrin, has been observed in fetal pancreatic cells, explaining the occurrence of pancreatic gastrinomas. There is interest in neutralizing gastrin’s effects through vaccination of patients with gastrointestinal tumors.9,10
Cholecystokinin
CCK was discovered as a factor that stimulated gallbladder contraction. It is secreted by open-type cells (I cells), which are most densely located in the small intestine and gradually decrease in number toward the large intestine. As a neurotransmitter, CCK is found in the central and peripheral nervous systems. The CCK gene is expressed only in rare neuroendocrine tumors and sarcomas. Measurement of circulating CCK by radioimmunoassay has been difficult for several reasons, including low blood concentrations (about 1 picomolar basally), the existence of several biologically active molecular forms, and cross-reactivity with gastrin.
CCK11 is the product of a 115-amino-acid precursor and has several molecular forms, of which the octapeptide of CCK (CCK-8), the eight C-terminal amino acids of CCK and the most abundant form of CCK in the CNS, is the most potent small peptide of CCK isolated. CCK-8, CCK-58, CCK-33, and CCK-39 have all been identified in significant amounts in human plasma. The differential processing of CCK in the CNS and gut is mediated by prohormone convertases (PCs). PC1 appears to be the major processing enzyme in the gut, while PC2 is more important in the brain. Peripherally, CCK acts through CCK1 receptors. CCK receptors have been detected in the gut, the pancreas, central and peripheral nervous systems, and lymphocytes. Several experimental CCK receptor antagonists are available.
The predominant stimulus to CCK release is the presence of breakdown products of fat and protein in the upper small intestine, specifically fatty acids of 10 to 18 carbon atoms, and aromatic-aliphatic amino acids. The CCK response is greater for unsaturated versus saturated fats. The mechanism by which CCK is released in response to nutrients appears to involve CCK-releasing factors.12 Bile salts may have an inhibitory effect on CCK release, since severe obstructive cholestasis or ingestion of cholestyramine increases plasma CCK levels. The bombesin family and adrenergic receptors stimulate CCK release.
The strongest evidence for the hormonal action of CCK is stimulation of gallbladder contraction, abolished by CCK1 receptor antagonists. Other CCK actions, such as pancreatic enzyme secretion, may be purely neural or have both neural and endocrine actions. CCK may have important trophic effects on normal and neoplastic tissues. CCK delays gastric emptying in several species, an effect that may be vagally mediated. Endogenous CCK may also play a role in gastric acid secretion and in the control of lower esophageal sphincter pressure. Other reported gastrointestinal actions of CCK include acceleration of intestinal transport, postprandial reduction in cephalically stimulated antroduodenal activity, and increased colonic transport. Exogenous CCK decreases meal size in animals and humans. No CCK excess clinical syndromes have been described. CCK has been linked to gastrointestinal motility disorders (e.g., constipation, irritable bowel syndrome), pancreatic disorders (pancreatitis, pancreatic tumor growth), and satiety disorders (obesity, anorexia). While CCK has been linked to these conditions, a pivotal role for CCK has not been unequivocally established.
Secretin
Secretin, the first hormone to be described, is secreted by upper duodenal and jejunal S cells.12–14 It is a 27-amino-acid linear peptide with a helical configuration. Secretin belongs to the secretin-glucagon family of peptides (see Table 152-1), and secretin receptors belong to the same family as CGRP, parathyroid hormone (PTH), and corticotropin-releasing hormone (CRH) receptors.
The major stimulus to secretin release is the presence of acid in the duodenum. Secretin is released when the pH of duodenal contents falls below 4.5; it has a circulating half-life of 2 to 3 minutes. Patients with achlorhydria have secretin deficiency, whereas hypersecretinemia occurs in Zollinger-Ellison syndrome. Fatty acids, bile salts, alcohol, and licorice also stimulate secretin secretion but to a lesser degree. Like CCK, secretin-releasing factors may be involved in the secretin response to luminal contents. Secretin release is inhibited by somatostatin and is suppressed in conditions associated with intestinal mucosal atrophy (e.g., celiac disease). Secretin is the principal hormonal stimulant of pancreatic and biliary water and bicarbonate secretion; CCK potentiates this effect. It may also contribute to pancreatic enzyme secretion.
Gastric Inhibitory Polypeptide
Gastric inhibitory polypeptide (GIP), also called glucose-dependent insulinotropic polypeptide, is a 42-amino-acid peptide secreted by mucosal K cells, predominantly found in the duodenum and jejunum and to a lesser extent in the ileum and colon. GIP is released into the circulation following a mixed meal and also ingestion of glucose, fat, or amino acids. Fat ingestion appears to be a more potent stimulator of GIP secretion than carbohydrate in humans. The peak concentrations are achieved after 30 to 60 minutes, although this is delayed up to 2 hours following fat ingestion. GIP is cleaved by the enzyme dipeptidyl peptidase IV (DPP-IV), which also hydrolyses glucagon-like peptide 1 (GLP-1) and peptide histidine methionine.
There are conflicting results regarding the role of GIP as a physiologic enterogasterone, but an important action of GIP is its glucoincretin role.15 The GIP response is absent and insulin response blunted in patients with the outdated jejunoileal bypass for morbid obesity. GIP has been implicated in the disordered insulin secretion of several diseases, including chronic pancreatitis, cirrhosis, and Turner’s syndrome. Secretion of GIP is enhanced in type 2 diabetes mellitus, but unlike GLP-1, there is little response to exogenous GIP. GIP has been reported to act on adipose tissue and may have an important role in lipid metabolism.16 It enhances fatty acid synthesis, lipoprotein lipase activity, and incorporation of free fatty acids into triglycerides. Several case reports of ACTH-independent, but food-dependent, Cushing’s syndrome have suggested that GIP may stimulate adrenal corticosteroid secretion through GIP receptors.17,18 GIP may also have a role in body weight regulation.
Peptide Products of Glucagon Gene
Glucagon is a 29-amino-acid residue peptide secreted by pancreatic islet alpha cells. It is a major hormone involved in glucose homeostasis. The mRNA of the glucagon precursor, preproglucagon, is expressed in the pancreas, intestine, and brain and gives rise to proglucagon, a 160-amino-acid residue peptide. Proglucagon is differentially processed in tissues (Fig. 152-4) such that it gives rise to glucagon, glicentin-related pancreatic peptide (GRPP), and a large major fragment in the pancreas. In intestinal L cells, it gives rise to GLP-1, GLP-2, and glicentin-enteroglucagon. Glicentin-enteroglucagon in turn gives rise to GRPP and oxyntomodulin, while GLP-1 is further processed to GLP-1(7-36) amide, GLP-1(9-36) amide (a GLP-1 antagonist), and GLP-1(7-37). Proglucagon in the hypothalamus is processed in a similar fashion to intestinal L cells.
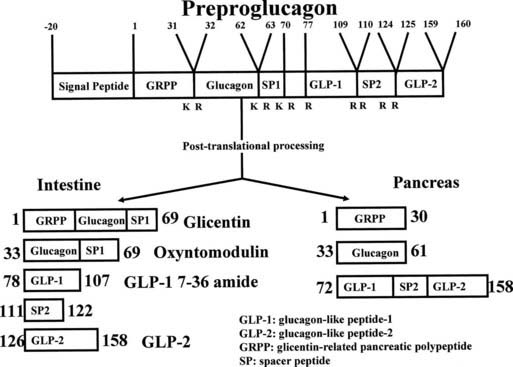
FIGURE 152-4. Posttranslational processing of proglucagon in the mammalian pancreatic α cell and small intestinal L cell. In the pancreas, the major products are glicentin-related pancreatic peptide, glucagon, and a C-terminal fragment containing the sequence of GLP-1 and GLP-2. In the L cell, the main products are glicentin (or enteroglucagon), oxyntomodulin, GLP-1 (7-36) amide, and GLP-2.
Proglucagon-derived peptides (PGDPs) are secreted by open-type L cells in the mucosa of the ileum and colon.19 A subpopulation of these cells also secretes PYY. PGDPs are secreted rapidly in response to mixed meals containing lipids and carbohydrates. Nonabsorbable sugars do not stimulate release of PGDPs. It is as yet unclear whether L cells are capable of sugar absorption and can couple absorption and secretion or receive signals from neighboring enterocytes. Several hormones and neurotransmitters, such as GIP, GRP, substance P, leptin, and neurokinin A, have been implicated in the regulation of PGDP release.
GLP-120 is a C-terminal amidated 30-amino-acid peptide and is the major incretin hormone, being the most potent insulinotropic hormone, greatly potentiating glucose-induced insulin secretion.20–23 The GLP-1 receptor antagonist, exendin(9-39) (derived from the venom of the Gila monster, Heloderma suspectum) reduces the insulin response to intestinal glucose. GLP-1 inhibits glucagon secretion indirectly through stimulation of somatostatin release. The effects of GLP-1 on glucagon and insulin secretion are glucose dependent, with little effect observed at glucose levels of (approximately) less than 3 mmol/L. Exogenous GLP-1 and its therapeutically used analogs can normalize blood glucose levels in type 2 diabetes mellitus, with significant reductions in HbA1c. GLP-1 has a half-life of only 4 minutes intravenously and 30 to 60 minutes subcutaneously. GLP-1 analog preparations (exenatide, liraglutide, taspoglutide, and others) used therapeutically have much longer half-lives. DPPIV inhibitors (gliptins) have also found a promising role in the treatment of diabetes.24
GLP-1 is an enterogastrone. It also inhibits pancreatic exocrine secretion, which may be secondary to its effect on gastric emptying. Gastric emptying is more rapid in obese patients who have reduced levels of postprandial GLP-1. GLP-1 almost completely inhibits vagally mediated (sham feeding–induced) gastric acid secretion, an effect that is completely lost after vagotomy. GLP-1 receptors in the circumventricular region of the brain may be responsible for this effect through central interaction with the autonomic nervous system. GLP-1 levels rise markedly in response to ileal infusion of nutrients, suggesting that GLP-1 may contribute to the ileal brake effect (i.e., endocrine inhibition of upper gastrointestinal motility and secretion elicited by the presence of nutrients in the distal small intestine). This may occur in combination with PYY, co-stored with GLP-1 in some intestinal L cells. Centrally, GLP-1 is a potent satiety factor.25,26
No physiologic actions of enteroglucagon or glicentin are known in humans. GLP-2, usually released in response to nutrients, is likely to be an important growth factor in the intestine. Circulating GLP-2 levels are elevated in response to gut injury. GLP-2 and its analogs may prove to be important in the treatment of intestinal injury.27,28 Exogenous GLP-2 has been reported to reduce bone resorption.29 Oxyntomodulin consists of the whole glucagon sequence extended at its C-terminus by a basic octapeptide. It inhibits gastric acid secretion and motility and has been shown to promote glucose production from hepatocytes. These effects occur at pharmacologic doses, suggesting that oxyntomodulin may act via receptors for GLP-1 and glucagon, at which it has weak agonist activity. Oxyntomodulin may also have a role in satiety and energy expenditure.30
Motilin
Motilin31 is a 22-amino-acid peptide found in open-type M cells, present in the epithelium throughout the small intestine but most dense in the duodenum and proximal jejunum. Motilin immunoreactivity is also found in the muscle layers of the intestine and has been detected in the large intestine, with the exception of the cecum, and in the gallbladder and biliary tract. Other gut hormones are released in response to the presence of nutrients in the gut, but motilin release in humans is not only influenced by ingestion of a meal (stimulated potently by fat intake, inhibited by carbohydrate) but also occurs in a cyclic manner during the interdigestive state. The cyclic release of motilin is associated with phase III of the migrating motor complex. Motilin induces phase III contractions in the stomach and associated phenomena, such as contraction of the lower esophageal sphincter, sphincter of Oddi, and gallbladder, and inhibition of gastric and pancreatic enzyme secretion. Atropine abolishes the cyclic release of motilin, suggesting a cholinergic regulatory mechanism, which in dogs appears to be through local nonvagal neurons. In dogs, plasma motilin levels are increased by bombesin, decreased by somatostatin (and perhaps pancreatic polypeptide), and abolished by food ingestion and insulin. Motilin appears to stimulate M cells, resulting in a positive-feedback loop. The motor effect of motilin on interdigestive motility is suppressed by a meal, duodenal acidification, somatostatin, pentagastrin, and CCK. The abolition of phase III contractions by atropine and 5-hydroxytryptamine antagonists suggests that cholinergic and serotoninergic mechanisms are substrates for motilin action. Other reported motilin actions vary among species and include effects on small-intestine water and salt absorption, colonic motility, gastric emptying, and pancreatic endocrine secretion. While the physiologic role of phase III contractions appears to be the prevention of the buildup of gut secretions (“housekeeping” role) and keeping the gut “warmed up” for the next meal, the physiologic role of motilin released in response to a meal is unknown. The prokinetic effect of macrolide antibiotics such as erythromycin, used in diabetic gastroparesis, is mediated via the motilin receptor. Other peptides with some homology to motilin include ghrelin. Ghrelin is unique in being a stomach-derived peptide that circulates in both acylated and nonacylated forms. Ghrelin is a potent appetite stimulator but has several other functions.31–33 A recently discovered product of the ghrelin gene is obestatin, a hormone whose physiologic role remains to be determined.34
Somatostatin
Somatostatin was first isolated from the ovine hypothalamus and exists as somatostatin-14 (S-14; 14 amino acids) and somatostatin-28 (S-28; 28 amino acids), which are cleavage products of the same precursor peptide.35–37 In the gut, somatostatin is found in endocrine D cells of gastric and duodenal epithelia, as well as extrinsic and intrinsic neurons of the intestinal myenteric and submucosal plexuses. S-14 predominates in gastric and pancreatic D cells, while the N-terminal extended S-28 predominates in intestinal mucosal cells. Somatostatin, which has a short half-life in the circulation, appears to function as a hormone and paracrine agent and as a neurotransmitter. It is released in response to a mixed meal. The mechanisms by which luminal stimuli result in somatostatin secretion are not fully understood. Other peptides that influence somatostatin release are gastrin, CCK, and bombesin, which are stimulatory, and the secretin family of peptides, which are inhibitory. Somatostatin release is stimulated by β-adrenergic agonists but inhibited by cholinergic agents.
Somatostatin is a universal inhibitor. Perhaps the best-described paracrine action of somatostatin is its inhibition of gastric acid secretion through its action on G cells, ECL cells, and parietal cells. Somatostatin also mediates the action of other enterogastrones on gastric function. Other inhibitory actions include effects on motility, intestinal and pancreatic secretion, release of gut peptides (including itself; autocrine), intestinal nutrient and electrolyte transport, growth and proliferation, and splanchnic blood flow. Five somatostatin G protein–coupled receptor subtypes (sst1-5), have been cloned, and multiple somatostatin receptor subtypes may be expressed by the same cell.35–37
Synthetic somatostatin analogs such as octreotide primarily act on sst2 receptors. These have been used in the treatment of several conditions, including neuroendocrine tumors and acromegaly (see Chapter 14). Longer-acting analogs and preparations have replaced octreotide, which has a short half-life and requires multiple daily subcutaneous injections. With longer-acting somatostatin analogs and preparations,38,39 however, breakthrough symptoms may occur and can be treated with the shorter-acting octreotide. Side effects of somatostatin-based treatments include malabsorption, steatorrhea, biliary sludge, and gallstone formation (usually asymptomatic). Radiolabeled octreotide scanning is routinely used for the localization of neuroendocrine and other tumors.40–42 Novel radiolabeled somatostatin analogs that bind a greater number of receptor subtypes than octreotide are currently being evaluated for the treatment of neuroendocrine and other tumors. The study of antineoplastic actions of somatostatin may prove useful in the treatment of a number of tumors.
Neurotensin
Neurotensin (NT) is a tridecapeptide. NT-related peptides include neuromedin N, xenopsin, and xenin. The highest concentrations of NT, released from N cells, have been observed in the ileal mucosa, with smaller amounts detected in the jejunum and duodenum. NT has also been detected in enteric neurons. The gut contains most of the body’s NT, with the remainder being mainly in the brain, particularly in the hypothalamus and pituitary. The most potent stimulator of NT is fat, and it has been proposed that NT mediates the gastrointestinal effects observed after fat ingestion, such as reduction in gastric emptying. Vagal and nonvagal mechanisms (e.g., neurally released bombesin) are involved in NT release. NT release is inhibited by somatostatin. Enhanced NT release is seen in patients suffering from dumping syndrome and after jejunoileal bypass. Elevated concentrations of NT have been detected in the plasma of patients with VIPomas.43
A number of biological actions have been attributed to NT and include vasodilation, increased histamine release from mast cells, inhibitory effects on gastric motility and gastric acid secretion, stimulation of the exocrine and endocrine pancreas, effects on intestinal motility and secretion, and inhibition of adipose-tissue blood flow. PP release invariably follows NT infusion; therefore NT may play a role in the enteropancreatic polypeptide axis. NT infusion stimulates glucagon but inhibits insulin release. NT may have growth-regulatory functions in the pancreas and colon, with its receptors detected in several cancer tissues.44
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


