FIGURE 145-1. Cartoon illustrating the cascade on transcription factor genes programming hypothalamic and pituitary embryogenesis and function. Most of this information was derived from genetic studies in mice. The list of genes and the complexity of the cascades increases progressively as basic and clinical research progress. In general, defects in early cascade genes produce nonviable or severe phenotypic syndromes in mouse and man. ACTH, Adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; FSH, follicle-stimulating hormone; GH, growth hormone; GnRH, gonadotropin-releasing hormone; GRH, growth hormone–releasing hormone; LH, luteinizing hormone; OT, oxytocin; PRL, prolactin; SS, somatostatin; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone; VP, vasopressin. See text and Refs. 10–20.
Growth Hormone and Prolactin
The human fetal pituitary gland can synthesize and secrete GH by 8 to 10 weeks of gestation.3,6 Pituitary GH content increases from about 1 nmol (20 ng) at 10 weeks to 45 nmol (1000 ng) at 16 weeks of gestation. Fetal plasma GH levels in cord blood samples are in the range of 1 to 4 nmol/L during the first trimester and increase to a mean peak of approximately 6 nmol/L at mid-gestation. Plasma GH levels fall progressively during the second half of gestation to a mean value of 1.5 nmol/L at term. Pituitary GH mRNA and GH content generally parallel the increase in plasma GH concentration between 16 and 24 weeks, reflecting a progressive maturation of hypothalamic-pituitary and forebrain function.6,7 The responses of plasma GH to somatostatin and GHRH and to insulin and arginine are mature at term in human infants.6,9
The high plasma GH concentrations at mid-gestation after the development of the pituitary portal vascular system may reflect unrestrained secretion. Studies in the sheep fetus have shown a failure of somatostatin to inhibit GHRH-stimulated GH release early in the third trimester, with maturation of the inhibitory effect of somatostatin near term.6,7 Studies of 9- to 16-week-old human fetal pituitary cells in culture have shown a predominant response to GHRH and a limited effect of somatostatin.21 In addition, there may be unrestrained pituitary GH secretory immaturity within the limbic and forebrain inhibitory circuitry that modulates hypothalamic function. Control of GH secretion matures progressively during the last half of gestation and the early weeks of postnatal life. Mature responses to sleep, glucose, and l-dopa are present by 3 months of age.
The pattern of ontogenesis of fetal plasma PRL differs significantly from that of GH; levels are low until 25 weeks and increase progressively thereafter to high levels at term.6 In vitro, fetal pituitary cells from mid-gestation fetuses show limited autonomous PRL secretion, although PRL release increases in response to TRH and decreases in response to dopamine.6,7 Estrogen stimulates PRL synthesis and release by pituitary cells, and the increase in fetal plasma PRL concentration in the last trimester parallels the increase in fetal plasma estrogen levels, although lagging by several weeks.9 Anencephalic fetuses have plasma PRL concentrations in the normal or low-normal range, supporting a role for estrogen in stimulating fetal PRL release. Studies in sheep demonstrate a similar pattern of fetal plasma PRL levels.6 These data support the view that maturation of brain and hypothalamic control of PRL, like GH, develops late in gestation and during the first months of extrauterine life.7,9,22
There is a general tendency toward hypersecretion of fetal pituitary hormones during the last half of gestation. Pituitary hormones found at high levels in cord blood from aborted human fetuses and premature human infants include GH, TSH, ACTH, β-endorphin, β-lipotropin, LH, and FSH.3,6,9 Development of hypothalamic-pituitary control involves maturational events in the cortex and midbrain, the hypothalamus and hypothalamic-pituitary portal vascular system, peripheral endocrine systems, and perhaps placental hormone and neuropeptide production. The fetal pituitary hypersecretion appears to be related to relatively delayed maturation of central nervous system and hypothalamic control of secretion of the hypothalamic releasing hormones.6
GH receptor mRNA levels and receptor binding are low in fetal liver, although receptor mRNA is present in other fetal tissues.3,6 The growth of anencephalic fetuses is nearly normal, however, suggesting that factors other than GH stimulate fetal IGF production. Nutritional factors are known to play a role.23,24 PRL receptors are present in most fetal tissues during the first trimester of gestation, and it is likely that lactogenic hormones have a significant role in organ and tissue development early in gestation.9,23 The coordinate increase in fetal adipose tissue and adipose-tissue PRL receptors, PRLR1 and PRLR2, suggests that PRL may play a role in growth and maturation of fetal adipose tissue later in gestation.3,25 PRL also may play a role in fetal skeletal maturation.25 Ovine placental lactogen stimulates glycogen synthesis in fetal ovine liver. Placental prolactins stimulate amino acid transport, DNA synthesis, and IGF-1 production in human fetal fibroblasts and muscle cells. GH and PRL have little activity in these tissues.9 (See Fetal Growth section.)
Fetal Pituitary-Adrenal Axis
The primordia of the adrenal glands can be recognized just cephalad of the bilaterally developing mesonephros by 3 to 4 weeks of gestation.3,26,27 The fetal adrenal is composed of three functional zones: a fetal zone capable of production of C19 androgens, a transitional zone with enzymes for cortisol production, and an outer definitive zone producing mineralocorticoids. The large eosinophilic cells of the fetal zone are well differentiated by 9 to 12 weeks of gestation and are capable of active steroidogenesis. The fetal adrenal gland grows rapidly and progressively to a combined glandular weight of approximately 8 g at term. Near term, the fetal zone makes up about 80% of the mass of the gland, with a relative size 10- to 20-fold that of the adult adrenal.3,26–28
Fetal adrenal cortical development is programmed by a cascade of genetic transcription factors. The major genes identified to date include Wt1, Sf1, and Dax129–31 (Fig. 145-2). Sf1 and Dax1 show coordinate expression in adrenal cortex, testis, ovary, hypothalamus, and pituitary tissues. Wt1 knockout mice have renal and gonadal abnormalities and lack adrenal glands.30 Sf1 gene knockout mice manifest adrenal and gonadal agenesis, gonadotropin deficiency, and absence of the hypothalamic ventromedial nucleus.30 Inactivating Dax1 gene mutations are associated with adrenal hypoplasia and gonadotropin deficiency. Several other transcription factors, notably Pbx1 and Wnt4, are involved earlier in the complex genetic cascade programming adrenal gland organogenesis from the coelomic epithelium and urogenital ridge.30,31 Pbx1 knockout mice die in utero with multiple organ defects that include adrenal agenesis and impaired testes development. Wnt4 gene description leads to abnormal adrenal development, masculinization of XX females, and müllerian-duct agenesis. The steroidogenic acute regulatory protein (StAR) is a rate-limiting factor in adrenal steroidogenesis.3,32 StAR knockout mice manifest glucocorticoid and mineralocorticoid deficiency and female genitalia in XY animals. In humans, inactivating StAR mutations cause adrenal hypoplasia and adrenal hormone insufficiency.32
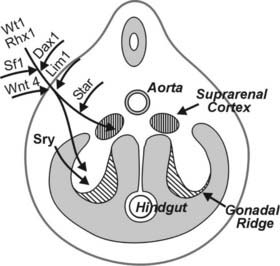
FIGURE 145-2. Cartoon showing a cross-section of a 5-week human embryo, with location of the adrenal primordia (suprarenal cortices) and gonadal ridges. The transcription factor genes known to be programming adrenal and gonadal embryogenesis are indicated. SF1 is required for testicular and ovarian development, while SRY is the single critical regulator of testicular embryogenesis. DAX1 gene inactivation leads to adrenal hypoplasia. The steroidogenic acute regulatory protein (StAR) is the rate-limiting factor for adrenal steroidogenesis. See text and Refs. 27–31.
Several growth factors also play an important role in adrenal development. Fibroblast growth factor (FGF) and epidermal growth factor (EGF) stimulate proliferation of both the fetal and definitive zones, and the fetal adrenal expresses high levels of IGF-2 mRNA and protein, which are responsive to ACTH.3,28 IGF-2 augments ACTH-stimulated expression of steroidogenic enzymes and stimulates steroid hormone production in fetal adrenal cortical cells. Cortisol production by the definitive zone does not occur de novo from cholesterol until 30 weeks of gestation, but some production using progesterone as precursor probably occurs earlier.3,28
The fetal adrenal expresses the same five steroidogenic apoenzymes as the adult gland: two microsomal enzymes with 17-hydroxylase and 17,20-desmolase (CYP17 or P450c17) and 21-hydroxylase (CYP21A2 or P450c21) activities, respectively; plus two mitochondrial cytochrome P450 enzymes providing cholesterol side-chain cleavage (CYP11A1 or P450scc) and C11/C18 hydroxylation of the parent steroid structure (CYP11B1/CYP11B2 or P450c11/aldosterone synthase).3,26,27 A fifth enzyme, expressed by the smooth endoplasmic reticulum, exhibits both 3β-hydroxysteroid dehydrogenase (3βHSD) and Δ4,Δ5-isomerase activities.26,27 The fetal zone has relatively high steroid sulfotransferase activity, and because of the low 3βHSD and high sulfotransferase activities, the major steroid products of the fetal adrenal are dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEAS), pregnenolone sulfate, several Δ5 3β-hydroxysteroids, and limited amounts of Δ5 3-ketosteroids, including cortisol and aldosterone.26,27 The definitive zone contributes only a small fraction of total fetal adrenal steroid output.
Much of the DHEA is converted to 16-hydroxy-DHEAS by fetal adrenal and liver. This programming is designed to provide DHEA substrate for placental estrone and estradiol production; 16-hydroxy-DHEA undergoes metabolism to estriol in the placenta. Fetal DHEAS production and maternal estriol concentrations increase progressively to term; DHEAS production approximates 200 mg/day near term.28 In the anencephalic fetus, placental estrogen production is reduced to about 10% of normal, and estrogen is known to suppress fetal zone growth of the fetal adrenal during the second half of primate pregnancy. It is proposed that estrogen feedback regulates secretion of fetal adrenal DHEA to maintain normal fetal-placental function and development.33
The major stimulus to fetal adrenal function is fetal pituitary ACTH.3,26,28,34 At mid-gestation, fetal plasma ACTH levels average 250 pg/mL, and although concentrations decline toward term, fetal ACTH levels exceed those later in life. Cholesterol, the major substrate for fetal adrenal steroidogenesis, is derived from circulating low-density lipoproteins (LDLs) and from de novo adrenal synthesis. LDL cholesterol, largely of fetal liver and testicular origin, contributes 70% of the total. Both fetal adrenal cortisol and placental estradiol regulate hepatic synthesis of cholesterol in the fetus.27 Both ACTH and angiotensin II receptors (AT1 and AT2) are present on fetal adrenal cells early in gestation. ACTH stimulates steroid production by activating StAR and increasing delivery of substrate cholesterol to P450scc; angiotensin II inhibits 3βHSD activity and promotes DHEA production in the fetal zone.26
Control of fetal ACTH secretion is complex.3 CRH protein has been demonstrated in fetal baboon pituitary, adrenal, liver, kidney, and lung tissues during the last third of gestation. Levels in pituitary are highest (300 to 500 pg/mg protein); levels in adrenal, lung, liver, and kidney tissues average 20 to 30 and 5 to 10 pg/mg protein, respectively.3,35 Circulating CRH levels are elevated in the fetus and probably result largely from extrahypothalamic and placental sources.35,36 Maternal levels of CRH are elevated during the last trimester of gestation and reach values of 0.5 to 1 nmol/L at term; normal values in nonpregnant women are less than 0.01 nmol/L.37 This placental CRH is bioactive, and levels correlate with maternal cortisol concentrations, suggesting that this circulating placental CRH plays a role in stimulating maternal corticotropin release. Fetal plasma CRH levels at term, however, are approximately 0.03 nmol/L and, relative to the presumably high levels in pituitary portal blood, probably have little role in modulating fetal corticotropin release. Mid-gestation fetal plasma ACTH concentrations average about 55 pmol/L (250 pg/mL), levels that maximally stimulate fetal adrenal steroidogenesis. Concentrations are higher throughout gestation than in postnatal life, although they fall near term.26,28 Although exogenous dexamethasone administered in pharmacologic doses to term infants is associated with short-term pituitary-adrenal suppression, this response is not noted in the fetus.38 Arginine vasopressin and catecholamines are also significant stimuli for fetal ACTH secretion.39
Near-term fetal cortisol production per unit body weight is similar to that in the adult.26 About two-thirds of fetal cortisol is derived from the fetal adrenal glands and one-third from placental transfer.26 The metabolic clearance of cortisol in the fetus is rapid; 80% is oxidized in fetal tissues or placenta to cortisone or further metabolites.26 Glucocorticoid receptors (GRs) are present at birth and are probably present at mid-gestation in most tissues, including placenta, lung, brain, liver, and gut.26,27,40–42 Mice lacking GR function manifest enlarged and disorganized adrenal cortices, adrenal medullary atrophy, lung hypoplasia, and defective gluconeogenesis.40
Fetal cortisol is converted to relatively inactive cortisone through an 11β-hydroxysteroid dehydrogenase (11βHSD) in fetal tissues; levels of circulating cortisone in the fetus at mid-gestation are four- to fivefold higher than cortisol concentrations (Fig. 145-3). This metabolism protects the anabolic milieu of the fetus because cortisol can retard both placental and fetal growth.43 As term approaches, selected fetal tissues, including liver and lung, express 11-ketosteroid reductase activity that promotes local conversion of cortisone to cortisol.26 An increase in fetal cortisol concentration occurs during the last 10 weeks of gestation and is the result of increased cortisol secretion and decreased conversion to cortisone.26 This increase in fetal cortisol production (the cortisol surge) has an important role in the maturation of several fetal systems or functions that are critical to extrauterine survival.26,44 (See Transition to Extrauterine Life.)
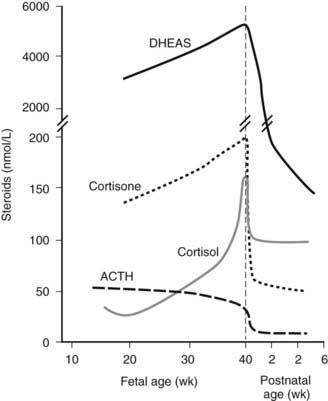
FIGURE 145-3. The pattern of maturation of serum cortisol, cortisone, and dehydroepiandrosterone (DHEA) in the human fetus during mid-gestation and the perinatal period. See text for details.
In developing mammalian fetal models, all components of the renin-angiotensin system, including renin, angiotensinogen, angiotensins, angiotensin-converting enzyme, and angiotensin II receptor, are present. The genes are generally expressed at higher levels in fetal than postnatal life.3,26 The human fetal adrenal gland is capable of aldosterone secretion near term, and fetal plasma aldosterone concentrations in infants who are born by cesarean section are three- to fourfold higher than maternal levels.26 Aldosterone secretion is low in the mid-gestation human fetal adrenal and is unresponsive to the secretagogues that are known to modulate aldosterone production in the adult. In sheep, fetal aldosterone becomes responsive to PRA and angiotensin II in the neonatal period.45,46 This situation also appears to be the case in the human fetus and neonate.
Mineralocorticoid receptors (MRs) are present in fetal tissues from 12 to 16 weeks of gestation.3,47 MR immunoreactivity is detectable in fetal kidney, skin, hair follicles, trachea and bronchioles, esophagus, stomach, small intestine, colon, and pancreatic exocrine ducts. The role of MRs in these fetal tissues remains unclear. MR knockout mice appear normal at birth but demonstrate defects in renin-angiotensin system functions in the postnatal period.48 Both subtypes of angiotensin receptors, AT1 and AT2, are detectable in various tissues early in fetal development.3,49 AT1 receptor mRNA expression in the fetal sheep kidney is low early in gestation, increases in the latter third of pregnancy, and decreases postnatally; AT2 mRNA levels, in contrast, are high at mid-gestation and decrease during the third trimester.49 These changes reflect growth factor–mediated changes in AT-containing cells in various tissues. Hormonal factors modulate fetal renal AT gene expression in sheep; angiotensin II suppresses both AT1 and AT2, and cortisol increases AT1 gene expression in kidney and lungs.49,50
The fetal renin-angiotensin system may have a role in maintaining renal excretion of salt and water into amniotic fluid to prevent oligohydramnios.3,46 The mechanism for the high aldosterone levels in the fetal and neonatal periods remains unclear. Atrial natriuretic peptide (ANP), a cardiac hormone, is known to inhibit aldosterone secretion. Because plasma ANP, brain (B-type) natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) concentrations are high in the fetus, the increased PRA and aldosterone levels are not due to relative natriuretic factor deficiency.51 Manifestations of mineralocorticoid deficiency in the newborn term infant can occur as a result of aldosterone deficiency or competition for binding to renal MRs by other steroids such as 17-hydroxyprogesterone.26 Relatively reduced glomerular filtration in the newborn limits sodium loss initially, but by 1 week of age aldosterone deficiency produces the characteristic manifestations of hyponatremia, hyperkalemia, and volume depletion.3,26
Pituitary-Gonadal Axis
The mammalian gonad is derived from two tissue anlagen: the primordial germ cells of the yolk-sac wall and somatic stromal cells that migrate from the primitive mesonephros.3,52,53 By 4 to 5 weeks of gestation, the germ cells begin migration from the yolk sac, and the gonadal ridge has appeared as a derivative of the mesonephros. The germ cells are incorporated into the developing gonadal ridge during the sixth week. Key regulators in the genetic cascade programming testes development include SRY, SF1 and DAX1.54,55 SRY is the single critical regulator. SF1 is required for testicular and perhaps ovarian development and mediates müllerian-inhibiting hormone gene expression and gonadotropin production. SF1 and DAX1 are orphan receptors of the steroid–thyroid hormone family of nuclear receptors involved in the regulation of target genes in the ventromedial hypothalamic nucleus, the hypothalamic gonadotroph cells, and the adrenal glands. The full menu of downstream gene targets remains to be defined, but a menu of prominent genes identified as programming gonadal development and sexual differentiation is shown in Fig. 145-4. Fetal pituitary gonadotropins are not required for gonadal development or sexual differentiation; LH or FSH receptor knockout mice are born phenotypically normal.56 Human mutations in several of the genes programming gonadal differentiation have been described.3,53 Loss-of-function mutations of SRY or Sox1 produces XY sex reversal, while gain-of-function mutations produce XX sex reversal. Sf1 mutations have been associated with XY sex reversal with gonadal agenesis. Wt1 mutations produce several syndromes associated with abnormal testicular embryogenesis (WAGR, Denys-Drash, and Frasier syndromes). Wnt4 gain-of-function mutations result in XX sex reversal, and Dax1 mutations produce SY sex reversal.53
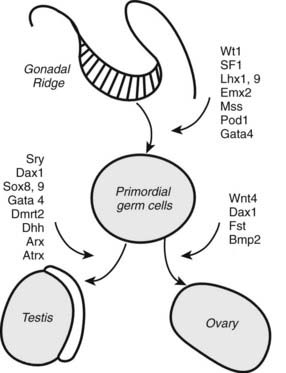
FIGURE 145-4. Cartoon showing the transcription-factor genes programming gonadal development. See text and Refs. 52–55.
Male gonadal differentiation begins at 7 weeks’ gestation with organization of the gonadal blastema into interstitium and germ cell–containing testicular cords.3,57 Leydig cells are visible by the end of the eighth week of gestation and are capable of androgen synthesis at this time. By 14 weeks, these cells make up as much as 50% of the cell mass. The fetal testes grow from approximately 20 mg at 14 weeks of gestation to 800 mg at birth; at 5 to 6 months they descend into the inguinal canal in association with the epididymis and the ductus deferens.57
In females, differentiation of ovaries begins during the seventh week of gestation.3,58 The gonadal blastema differentiates into interstitium and medullary cords containing the primitive germ cells now referred to as oogonia. The cords degenerate, and cortical layers of surface epithelium containing individual small oogonia appear. By 11 to 12 weeks, clusters of dividing oogonia are surrounded by cord cells within the cortex.58 Primordial follicles appear at about 18 weeks, and the number increases rapidly thereafter.59 The number of oocytes progressively declines from a peak of 3 to 6 million at 5 months to some 2 million at term.28,59 Germ cell proliferation and apoptosis are ongoing simultaneously; only those oocytes enfolded by developing granulosa cells (as primordial follicles) survive.28,59 By 5 to 7 months, stroma-derived thecal cells develop around the primordial follicles as they mature to primary follicles. Each fetal ovary weighs about 15 mg at 14 weeks of gestation and 300 to 350 mg at birth.58 Interstitial steroid-producing cells are present after 12 weeks, and during the third trimester, theca cells with steroidogenic capacity surround the developing follicles.28
Estrogen effects are mediated by two receptors, estrogen receptors α and β (ERα and ERβ).3,60,61 The mRNAs for both receptors have been characterized in the 16- to 23-week human fetus. One or both receptor mRNAs are present in most tissues. ERβ message is predominant, particularly in testis, ovary, spleen, thymus, adrenal, brain, kidney, and skin. ERα message is prominent in uterus, with relatively low levels in most other tissues.60,61 The significance of estrogen receptors in fetal development remains unclear. Knockout of the ERα gene in mice does not impair fetal development of any tissue; but adult females are infertile, with hypoplastic uteri and polycystic ovaries; adult males manifest decreased fertility.61 ERβ knockout mice develop normally, and female adults are fertile with normal sexual behavior; adult males reproduce normally but have prostate and bladder hyperplasia.60 Knockout of both ERα and ERβ genes also has little impact on fetal development, but after birth the uterus, fallopian tubes, vagina, and cervix in females are hypoplastic and unresponsive to estrogen.61
In the male fetus, there is an increase in testicular testosterone production stimulated by the high levels of circulating placental human chorionic gonadotropin (hCG) between 10 and 20 weeks.62,63 Fetal pituitary LH contributes to fetal Leydig-cell stimulation, but the predominant gonadotropic effect at this time is hCG. Fetal testosterone stimulates male sexual differentiation of the primitive mesonephric ducts (to form the ductus deferens, epididymis, seminal vesicles, and ejaculatory ducts) and masculinizes the urogenital sinus and external genitalia (to form the prostate gland, phallus, and penile urethra).
Androgen receptors appear in the mesenchyme of urogenital structures at 8 weeks of gestation, followed by appearance of the receptors in the epithelium at 9 to 12 weeks.3,64 There is no difference in receptor expression in male and female fetuses. Dihydrotestosterone (DHT) stimulates male differentiation of the urogenital sinus and external genitalia, including differentiation of the prostate, growth of the genital tubercle to form a phallus, and fusion of the urogenital folds to form the penile urethra. DHT is formed from testosterone by the 5α-reductase enzyme within the urogenital sinus and urogenital tubercle and acts through the same androgen receptor that mediates the action of testosterone in the wolffian ducts.
Sertoli cells in the male gonad produce müllerian-inhibiting substance (MIS), which dedifferentiates the primitive müllerian duct system.65–67 MIS, a 12-kD glycoprotein, is a member of the transforming growth factor β (TGF-β) family. MIH gene expression, stimulated by SRY, continues throughout gestation at the time of müllerian duct regression and then decreases after birth.68 MIH also may facilitate testicular descent. In the female fetus, müllerian duct differentiation proceeds in the absence of MIH, and the primitive mesonephric duct system dedifferentiates in the absence of testosterone. In the absence of dihydrotestosterone, the urogenital sinus and external genitalia differentiate to the female phenotype.
Both androgens and estrogens are involved in the structural development of the rodent brain.3,69–71 Gonadal hormones control gonadotropin production in the hypothalamus, programming cyclic ovarian function and function of the testes.70,71 Testosterone administration to neonatal female rats produces permanent inhibition of cyclic hypothalamic control through local aromatization to estradiol. In primates and humans, estrogens seem to be more effective in this regard. However, there is no evidence for permanent programming in the primate, and there appear to be no major tissue biochemical differences between the sexes in utero to account for sexual dimorphic behavioral or gonadotropic programming.72
Pituitary-Thyroid Axis
The thyroid gland is a derivative of the primitive buccopharyngeal cavity and is composed of two sets of anlagen: a median anlage derived from the pharyngeal floor and paired lateral anlagen from the fourth pharyngeal pouch.73,74 At least five transcription-factor genes in mice are involved in programming thyroid gland development from these anlagen, including genes coding for HEX, EYA1, thyroid transcription factor 1 (TTF1), TTF2, and PAX874–78 (Fig. 145-5). Hex gene knockout in mice is associated with thyroid agenesis or severe hypoplasia. Eya1 gene knockout is associated with thymus and parathyroid agenesis and thyroid hypoplasia. Knockout of the gene coding for TTF1 results in pulmonary hypoplasia and thyroid agenesis. Knockout of the mouse gene for TTF2 results in thyroid dysgenesis and cleft palate, whereas mutation of the gene coding for PAX8 results in thyroid gland hypoplasia and renal anomalies.74–78 Pharyngeal pouch-derived parafollicular C cells are aplastic in the Ttf1 knockout mice but normal in the Pax8 knockouts.79 Optimal expression of differentiated postnatal follicular cell function, including normal thyroglobulin, thyroid peroxidase, and TSH receptor activities, requires all three gene effects.75 Other gene activities, including endothelin and TSH receptor (TSHR) activities, may also play a role in thyroid gland differentiation.76,80 Gene mutations for TTF1, TTF2, PAX8, and TSHR have been described in human neonates with thyroid dysgenesis, but they account for less than 10% of cases.76
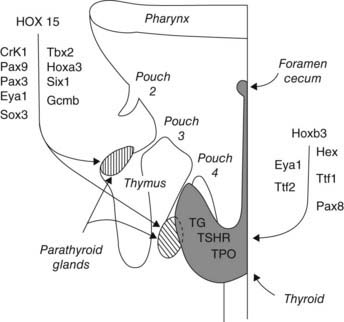
FIGURE 145-5. Cartoon showing the homeobox genes programming development of the thyroid and parathyroid glands. Most of this information was derived from genetic studies in mice. See text and Refs. 74, 75, 150, 152, and 153.
Thyroid gland embryogenesis is largely complete by 10 weeks of gestation, at which time colloid formation is demonstrable; by 12 weeks, the gland weighs about 80 mg. At term gestation, gland weight approximates 1 g.3,81 Pituitary and plasma TSH concentrations increase during the second trimester in the human fetus, coincident with development of the pituitary-portal circulation.
Pituitary and plasma thyrotropin (TSH) concentrations begin to increase during the second trimester in the human fetus, about the time that pituitary-portal vascular continuity develops.3,81–83 TSH levels increase progressively during the last half of gestation. Plasma thyroid hormone–binding globulin (TBG) and total T4 concentrations increase progressively from low levels at 16 to 18 weeks of gestation to maximal levels at 35 to 40 weeks. Free T4 levels also increase as a consequence of the increase in T4 production. The increases in circulating TSH and T4 levels during the third trimester reflect a progressive maturation of hypothalamic-pituitary control and thyroid gland responsiveness to TSH. Negative feedback hypothalamic-pituitary-thyroid control of TRH and TSH secretion develops during the latter half of gestation, and hyperthyrotropinemia in response to hypothyroidism in utero is manifest by 22 to 24 weeks’ gestation.
The period of parallel increases in fetal TSH and free T4 levels during the latter half of gestation is followed by the sequential TSH, free T4, and T3 surges in the early neonatal period and a final slow equilibration of the TSH/free T4 ratio to adult values during infancy and childhood.84,85 This maturation requires coordinate maturation of hypothalamic TRH secretion, pituitary TRH sensitivity, thyroid follicular-cell responsiveness to TSH, and thyrotropin negative-feedback modulation of TSH secretion. Functionally, the fetus progresses from a state of both primary (thyroidal) and tertiary (hypothalamic) hypothyroidism at mid-gestation, through a state of mild tertiary hypothyroidism during the final weeks in utero, to a fully mature hypothalamic-pituitary-thyroid axis by about 2 months postnatally.3,85 Premature infants born at 26 to 28 weeks’ gestation respond to exogenous TRH with an increase in plasma TSH comparable to that in adults.84 However, their TSH (surge) response to extrauterine exposure is obtunded, and their T4 and T3 increments are reduced relative to term infants.86 These responses are further attenuated at 28 to 30 weeks and absent in 23- to 27-week infants, reflecting a relative hypothalamic-pituitary-thyroid system immaturity inversely correlated in severity with gestational age.86
T4 can be monodeiodinated to either to active 3′,3,5-triiodothyronine (T3) or inactive 3,3′,5′-triiodothyronine (reverse T3 [rT3]) by one of the three iodothyronine monodeiodination enzymes, type 1, type 2, or type 3.81–83 Deiodination of the outer (phenolic) ring of T4 to form T3 is catalyzed by either monodeiodinase 1 (MDI-1) or MDI-2. MDI-1 is principally expressed in fetal hepatic and renal tissues, whereas MDI-2 is expressed in the placenta and in the fetal brain, pituitary, and brown adipose tissues. MDI-3 in fetal placenta, brain and skin catalyzes deiodination of the inner (tyrosyl) ring of T4 to produce inactive rT3.82 Fetal thyroid hormone metabolism is characterized by low levels of MDI-1 and high levels of MDI-3 activity.82,83 Thus in the fetus, T4 is largely converted to rT3, and serum rT3 levels during the third trimester are high, ranging from 2 to 4 nmol/L. Fetal serum T3 levels remain low until the final weeks of gestation. Plasma T3 levels are less than 0.2 nmol/L before 30 weeks’ gestation and then increase slowly to levels averaging 0.7 nmol/L at term81,82 (Fig. 145-6). The low levels of circulating T3 in the fetus are due to relatively decreased production and increased degradation via MDI-3. Early in gestation, placental transfer is the only source of T4 in fetal fluids and is essential for normal fetal neurodevelopment; T4 is detectable in human coelomic fluid at levels of 0.5 to 2 nmol/L between 6 and 11 weeks of gestation, before the onset of fetal thyroid function.87 Significant placental transfer continues to term, when serum T4 levels in the athyroid fetus range from 30 to 70 nmol/L (2.3 to 5.4 µg/dL).88 Isotopic equilibrium studies with pregnant rats at term suggest that 15% to 20% of the T4 in fetal tissues is of maternal origin.89
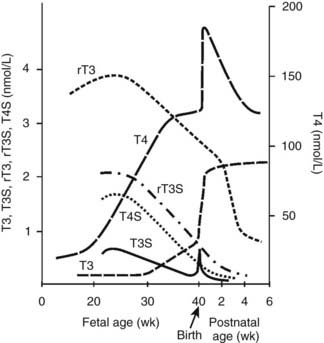
FIGURE 145-6. The pattern of maturation of thyroid hormone levels in the human fetus from mid-gestation through the perinatal period. The TSH surge (not shown) at birth and the prenatal increase in fetal hepatic monodeiodinase (MDI) type 1 activity stimulate marked increases in T3 and T4, facilitating the transition to postnatal life. The levels of bioinactive sulfated iodothyronine analogues T3S, T4S, reverse T3 (rT3)S, and T2S (not shown) peak during mid-gestation and fall progressively to term. See text (Pituitary-Thyroid Axis and Adaptation to Extrauterine Life) for details.
Fetal thyroid hormone metabolism is characterized by predominant conversion of T4 to inactive rT3 and sulfated analogues. The production of T3 is minimal until the last few weeks of gestation. The predominant production of inactive thyroid metabolites serves to potentiate the fetal anabolic milieu. The sulfated iodothyronine analogues are major thyroid hormone metabolites in the fetus.82,90 They include T4S, T3S, rT3S, diiodo- and monoiodothyronine sulfates, and triiodothyroacetic acid (TRIAC) sulfate.90,91 These analogues serve as substrate for MDI-1 and accumulate in serum as a result of increased production in fetal liver and relatively low hepatic MDI-1 activity. Concentrations peak near mid-gestation (see Fig. 145-6) because of increased production and decreased degradation; the sulfate metabolites are not substrates for MDI-3. Reverse T3 and the sulfated analogues in fetal sheep in the third trimester of gestation account for more than 90% of T4 metabolites.82,90 Relatively high levels of MDI-2 activity are expressed in fetal brain and brown adipose tissue at mid-gestation (before the near term increase in plasma T3 levels) and provide for local tissue production of T3 during the latter half of gestation.82,90 Although fetal thyroid development occurs largely independent of maternal influences, significant maternal-to-fetal transport of thyroid hormones occurs and is estimated to supply 20% to 30% of fetal hormone turnover at term.82,92 This T4 of maternal origin may serve as an important substrate for local T3 production by MDI-2-expressing tissues (e.g., brain) in the hypothyroid fetus.81,82
The adult thyroid follicular cell can modify iodine transport or uptake with changes in dietary iodine intake, independent of variations in serum thyrotropin levels.3,93,94 Before 36 to 40 weeks’ gestation, the thyroid gland lacks this autoregulatory mechanism and is susceptible to iodine-induced inhibition of thyroid hormone synthesis. The fetal thyroid cell, when exposed to high circulating levels of iodide, is unable to reduce iodide trapping and prevent the high intracellular iodide concentrations that produce the blockade of hormone synthesis referred to as the Wolff-Chaikoff effect. Failure of the immature thyroid to exhibit autoregulation is due to failure of down-regulation of thyroid cell membrane sodium-iodide symporter units.93
Several classes of cell membrane iodothyronine transporters have been recently described.3,95,96 These transporters belong to several families of organic anion, amino acid, and monocarboxylate solute carriers, including the anion transporting polypeptide (OATP) family and the solute carrier family 21 (SLC21).95,96 The significance of these transporters is not yet clear, but mutation of the human monocarboxylate transporter 8 (MCT8), a member of the SLC21 family, a specific thyroid hormone transporter present in developing brain, leads to a syndrome of combined thyroid dysfunction and psychomotor retardation.96 MCT8 expression in neonatal mice has been localized to neurons in the olfactory bulb, cerebral cortex, hippocampus and amygdala. Presumably all thyroid hormone–sensitive cell populations express iodothyronine membrane transporters. A cell surface αVβ3 integrin T4 receptor mediates T4-induced activation of the MAPK pathway for angiogenesis and perhaps actin polymerization and neuronal migration.97
Classical thyroid hormone actions are mediated via functional thyroid hormone nuclear receptors. Two genes code for the receptors, TRα on chromosome 17 and TRβ on chromosome 3.3,98 The genes code for four classical receptor isoforms TRα1, TRα2, TRβ1, and TRβ2, which bind thyroid hormones and bind to DNA to effect gene transcription. TRα2 does not bind thyroid hormone but binds to DNA and can inhibit binding of other TRs. The TRs exist as monomers, homodimers and heterodimers with other nuclear receptor family members such as retinoid X (RXR). Other TR transcripts including TRΔα1 and TRΔα2 have been characterized. These do not bind DNA or T3 but can inhibit TR and retinoid receptor activities.99 The TR are expressed developmentally and differentially in various fetal and adult tissues. TRα proteins are present in most tissues. TRβ1 is expressed in liver, kidney, and lung and in developing brain, cochlea, and pituitary. TRβ2 expression is restricted largely to the pituitary gland, retina, and cochlea.98 The receptors function redundantly, as indicated by knockout studies in mice, but predominant effects of one or another TR have been characterized. Knockout of both the TRα and TRβ genes in mice is not lethal but results in elevated TSH levels, deafness, bradycardia, and decreased postnatal growth with delayed bone maturation.98,99
In the fetal rat brain, TRα1 mRNA and receptor binding are detectable by 12 to 14 days gestation (term is 21 days), increasing to maximal levels at birth. The TRβ1 isoform is detected at birth and increases some 40-fold in the early postnatal period.100 In human fetal brain, TRα1 and TRβ1 isoforms and receptor binding are present by 8 to 10 weeks’ gestation; TRα1 transcripts and receptor occupancy increase 8- to 10-fold by 16 to 18 weeks.101,102 Liver, heart, and lung receptor binding can be identified by 13 to 18 weeks.102,103
The timing of thyroid hormone-programmed development of fetal tissues is selective and requires the interaction of local tissue MDI-1, thyroid receptors, receptor coactivators, and thyroid-responsive genes.3,99,104–108 The type and timing of tissue MDI determines the availability of T3.104,105 In the absence of T3, the unliganded receptor (aporeceptor) recruits corepressors, repressing gene transcription; non-T3 binding receptors also can repress transcription and tissue maturation by inhibiting receptor DNA binding.106–108
In hypothyroid mice, repressive effects of aporeceptors have been shown to delay tissue maturation in brain, bone, intestine, spleen, and heart.108 The timing of tissue maturation events in the mouse range from early midbrain development at gestational day 15 (roughly equivalent to mid-gestation in the human fetus) through perinatal hypothalamic maturation and activation of hepatic enzymes, cardiac ion channels, and spleen erythropoiesis to postnatal brain, intestinal, and bone maturation and thermogenesis.3,104–111 In the human infant, as in the mouse, the increase in circulating T3 levels associated with parturition normally triggers maturation of tissue and organ functions essential to postnatal metabolism and homeostasis (e.g., hepatic, intestinal and cardiac functions and brown-fat thermogenesis). Thyroid hormone–stimulated maturation of vision and hearing appear to be triggered by the local expression of MDI-2 mediating local T3 production, postnatally in the mouse and probably toward the end of the second trimester in the human fetus.111
Classical signs of congenital hypothyroidism (jaundice, lethargy, feeding difficulties, macroglossia myxedema, hypothermia, growth retardation, progressive developmental delay, and IQ deterioration) accrue during the early weeks and months of extrauterine life as maternal T4 becomes unavailable and the non-CNS tissues become thyroid hormone responsive.82 Maternal hypothyroxinemia during pregnancy has been associated with attention deficit disorder and/or 5 to 10 points of IQ deficit in the offspring of such pregnancies.100 The period of brain dependency for thyroid hormone extends postnatally to 2 to 3 years of age, but the early weeks and months of life are most critical. Untreated thyroid agenesis is associated with a loss of 5 to 7 IQ points monthly during the first months of postnatal life and over 6 to 8 months can amount to a 30- to 40-point IQ deficit.83
THE HYPOTHALAMIC–POSTERIOR PITUITARY SYSTEM
Arginine vasopressin is detectable after 10 to 12 weeks of gestation in the human fetal neurohypophysis. By 40 weeks, the level approximates 20% of that in adults. Fetal pituitary oxytocin levels, detectable by 11 to 15 weeks, exceed AVP concentrations by 19 weeks. The AVP-oxytocin ratio falls progressively thereafter. At 11 to 19 weeks of gestation, immunoreactive arginine vasotocin (AVT), characteristic of submammalian species, is found in the human fetal pituitary at approximately two-thirds the concentration of AVP and is secreted by cultured human fetal pineal cells during the second trimester.3,112,113 In addition, AVT is found in mammalian cerebrospinal fluid, fetal pituitary, and adult pineal gland but not in the adult pituitary. In adult mammals, instillation of AVT into cerebrospinal fluid inhibits gonadotropin and corticotrophin release, stimulates PRL release by the anterior pituitary, and induces sleep; however, its physiologic importance in these regards remains unclear.113 The role of AVT in the fetal pineal gland is unknown. The transcription factor genes programming development of the neurohypophysis are shown in Fig. 145-1.
The neurohypophyseal peptides are synthesized as large precursor molecules (neurophysins) and processed to bioactive amidated peptides.3,114 Enzymatic processing involves progressive cleavage of carboxyl terminal–extended peptides producing sequentially (for OT) OT-glycine-lysine-arginine (OTGKR), OTGK, OTG, and OT. Similar progressive processing yields AVPG and AVP from the AVP neurophysin. Enzymatic processing of neurophysins matures progressively in the fetus so that early in gestation, fetal plasma contains relatively large concentrations of the extended peptides. For OT, the ratio of OT extended peptides to OT in fetal sheep serum is approximately 35 : 1 early in gestation and 3 : 1 late in gestation.114 Aquaporin-1, aquaporin-2, and aquaporin-3 water channel receptors are present in human fetal and newborn kidney, and the ability of the newborn infant to respond to isotonic dextran or hypertonic saline with appropriate alterations in kidney free-water clearance indicates that human volume and osmolar control systems for AVP secretion are mature at birth.115,116
In the fetal sheep, the baseline fetal plasma AVP concentrations are similar to maternal levels after mid-gestation. During the last trimester of gestation, fetal hypothalamic and pituitary responsiveness to both volume and osmolar stimuli for AVP secretion are well developed, and AVP exerts antidiuretic effects on the fetal kidney.112 Baseline plasma levels of AVT in fetal sheep during the last trimester approximate values for AVP and OT.113 Presumably this AVT is derived from the posterior pituitary, but the stimuli for AVT secretion in the fetus are not defined.
During the final third of gestation, the sheep fetus responds to hemorrhage, hypertonic saline, angiotensin II, and hypoxia with increased AVP secretion. The inverse relationship of AVP mRNA and pituitary AVP content in the near-term fetal sheep suggests a dynamic AVP synthesis–content feedback relationship.112,117 The responses to hypertonicity and dehydration in the newborn lamb are quantitatively comparable to the adult responses, whereas the response to hypoxia greatly exceeds that of the adult.112
In the fetus, AVP appears to function as a stress-responsive hormone. Perhaps the major potential stress for the fetus is hypoxia, and the response of AVP to hypoxia is increased compared with the maternal response and with the fetal AVP responses to osmolar stimuli.3,118–121 Plasma AVP concentrations in human cord blood are elevated in association with intrauterine bradycardia and meconium passage.120 The vasopressor action of AVP may be important in the maintenance of fetal circulatory homeostasis during hemorrhage and hypoxia; AVP has a limited effect on fetoplacental blood flow.121,122 Fetal hypoxia is also a major stimulus for catecholamine release. There is little information on interaction between AVP and catecholamines during fetal hypoxia, but both fetal hypoxia and AVP stimulate anterior pituitary function.121 A role for AVP as a CRH is established in the adult, and the ovine fetal pituitary responds separately and synergistically to AVP and CRH early in the third trimester.123 The role of AVP in controlling fetal corticotrophin release seems to decrease with gestational age. It is not known whether AVT functions as a fetal CRH.
OT receptors have been demonstrated in human fetal membranes at term, and AVP receptors have been found in renal medullary membranes of newborn sheep.3,124–126 Both AVP and AVT evoke antidiuretic actions in the sheep fetus during the last third of gestation, and both hormones act to conserve water for the fetus by inhibiting fluid loss into amniotic fluid through the lungs and kidneys.118,122 Whether AVT exerts its effects through AVP receptors or separate fetal AVT receptors is not clear. Maximal concentrating capacity by the fetal kidney is limited to about 600 mmol/L. This limitation is due not to adequate AVP stimulation but rather to inherent immaturity of the renal tubules.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


