FIGURE 120-1. Timeline of changes in the reproductive axis.
As puberty progresses, there is little change in the frequency of LH pulsations. However, the mass of hormone secreted during each pulse and the amplitude of each pulse increase strikingly,8 suggesting that the sexual-maturation change in LH consists of a specific mass/amplitude-dependent mechanism. Because the metabolic clearance rate of FSH is much slower than that of LH, it is more difficult to characterize the pulsation characteristics of FSH. However, GnRH induces the release of both LH and FSH, and cross-correlation studies confirm that LH and FSH secretory bursts occur simultaneously at all ages. In boys studied longitudinally, the percentage of peaks with high pulse amplitude increases steadily with each stage of puberty9 (Fig. 120-2). In early puberty, there is a nocturnal increase in LH, suggesting a sleep-associated regulatory mechanism that either activates or de-represses pathways controlling GnRH release. At the completion of puberty, there is no day/night difference in LH/FSH pulsations.8,9 Thus the nocturnal activity of the GnRH pulse generator appears to represent a discrete developmental stage. In patients with anorexia nervosa who develop hypogonadotropic hypogonadism but then gain weight, recovery of the reproductive axis is heralded by nocturnal increases in gonadotropins,10 suggesting a reactivation of the GnRH pulse generator rather than an age-specific event. The pubertal rise in mean LH and FSH stimulates the ovaries to secrete increasing amounts of estradiol and the testes to secrete increasing amounts of testosterone. These steroids then stimulate the development of secondary sex characteristics.
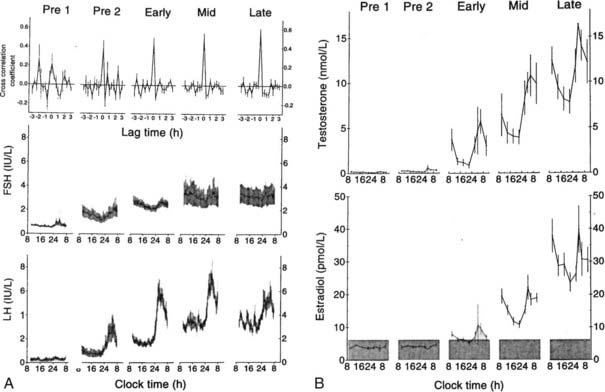
FIGURE 120-2. A, Mean luteinizing hormone (LH) and follicle-stimulating hormone (FSH) measured each 20 minutes for 24 hours in 12 boys monitored throughout puberty. Pre 1 and Pre 2 designate two prepubertal times. Pre 1 had testicular volumes of 1 to 2 mL; Pre 2 had testicular volumes of 2 mL. Early, mid, and late designate early puberty, midpuberty, and late puberty. B, Mean 24-hour blood testosterone and estradiol in the same 12 subjects.
(From Albertsson-Wikland K, Rosberg S, Lawering B et al: Twenty-four-hour profiles of luteinizing hormone, follicle-stimulating hormone, testosterone, and estradiol levels: a semilongitudinal study throughout puberty in healthy boys, J Clin Endocrinol Metab 82:541–549, 1997. © The Endocrine Society.)
In addition to these changes in LH, FSH, and gonadal steroids, gonadal production of inhibin A and B changes during puberty. The chemistry and function of the inhibins and activins are described separately in Chapter 116 (Burger). Girls have higher mean FSH, particularly during puberty stages II to IV, than boys.11 Inhibin is produced by the Sertoli cells of the testis and the granulosa/theca cells of the ovary (see later discussion). Inhibin B is the physiologically important form in men.12 Serum levels of inhibin B increase between stages I and II of puberty and remain constant from stage II to stage V. This finding is in contrast to FSH concentrations, which increase to stage III and remain constant thereafter. In late puberty (stage II on), inhibin B is negatively correlated with FSH, a phenomenon that is also present in adult men. Inhibin production by the ovary varies during the menstrual cycle.13 Inhibin B increases during the follicular phase, whereas inhibin A increases during the luteal phase. Inhibin B has been used as an index of ovarian reserve, perhaps reflecting the pool of primordial follicles.14
TRIGGERS FOR THE ONSET OF PUBERTY
The physiologic mechanisms controlling the onset of puberty remain an important unsolved problem in endocrinology. As noted earlier, the onset of puberty is perhaps more accurately described as reactivation of the reproductive axis. Although the regulatory mechanisms that control puberty are incompletely understood, genetic disorders provide important clues to the critical steps.15,16 Mutations have been identified in: (1) the pathway that controls the development or function of GnRH-producing neurons (KAL1, FGFR1, GPR54, NELF, PROK2, PROKR2); (2) transcription factors that regulate normal development of the ventromedial hypothalamus and pituitary (SF1, DAX1, HESX1, LHX3, SOX2 or PROP1); (3) GNRH1 and GnRH-R; and (4) the gonadotropin genes (LHβ or FSHβ, SF1, DAX1). Genes that affect hypothalamic regulation of GnRH secretion (LEP, LEPR) can also cause inherited hypogonadotropic hypogonadism. Thus, each of these steps is necessary for GnRH stimulation of gonadotropin secretion. However, there is no evidence that changes in the expression or regulation of these genes is the key regulatory step for activation of puberty.
GPR54 and its ligand, kisspeptin, are good candidates as key regulators of the onset of puberty. GPR54 was discovered based on linkage studies of subjects with idiopathic hypogonadotropic hypogonadism without anosmia or other features associated with Kallmann’s syndrome.17,18 GPR54 encodes a G protein–coupled receptor that binds kisspeptin, a protein expressed in hypothalamic neurons co-located with GnRH neurons.19 Targeted mutagenesis of the GPR54 receptor in mice causes gonadotropin deficiency and failure to undergo secondary sexual maturation.18 The GPR54 knockout mice have apparently normal GnRH-producing neurons and respond to exogenous GnRH by producing pituitary gonadotropins. Thus, pituitary function is normal, and the defect appears to involve an upstream pathway that regulates GnRH release. Administration of kisspeptin stimulates GnRH secretion in mice20 and in prepubertal primates.21 Further studies of this pathway are needed, but these findings suggest that kisspeptin or related ligands, acting via GPR54, may be the proximate pathway for reactivating the GnRH pulse generator and regulating the onset of puberty. If correct, this idea begs the question, what regulates production of kisspeptin and GPR54? Moreover, it is important to integrate other physiologic events that occur at the time of puberty.
One of the most important physiologic factors regulating the onset of puberty appears to be energy balance. Indicators of this status, such as leptin and ghrelin, have been shown to affect gonadotropin secretion. Leptin signals that adequate fat mass is available before entering the reproductive phase.22 In leptin-deficient patients, there is initiation or resumption of gonadotropin secretion after leptin replacement.22 Recombinant leptin restores gonadotropin secretion in women with hypothalamic amenorrhea associated with anorexia nervosa.23 Leptin levels rise by 50% just before the onset of puberty and then fall to baseline after stage II.24,25 Thus, leptin is necessary for activation of the GnRH pulse generator. GnRH neurons, however, do not express leptin receptors, and there is no compelling evidence that it is the primary trigger for the onset of puberty.26 Ghrelin, the ligand of the growth hormone secretagogue receptor, also plays a role in signaling energy status, although this effect seems to differ between males and females. Ghrelin is secreted in response to food deprivation. Prepubertal male rats treated with ghrelin showed decreased serum LH and testosterone levels and delayed puberty. Females, on the other hand, showed no changes in gonadotropin secretion or pubertal timing.27 These data suggest that sex differences may add an additional layer of complexity to the timing of puberty. Sleep is another physiologic factor implicated in the regulation of puberty. The initial nocturnal GnRH surges suggest a link to circadian pathways, or possibly disinhibition during REM sleep. Thus, multiple pathways may converge to signal the onset of puberty; key steps involve activation of GPR54, adequate energy balance, and activation of the GnRH pulse generator. The interaction of the genes, gonadotropins, and energy balance are summarized in Fig. 120-3.
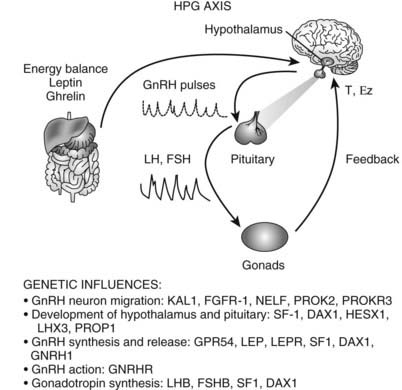
FIGURE 120-3. Triggers for the onset of puberty. Genes associated with the onset of puberty are listed below the steps they regulate. KAL1, Kallmann syndrome gene 1 (encodes anosmin 1); FGFR1, fibroblast growth factor receptor 1; NELF, nasal embryonic luteinizing hormone releasing hormone factor; PROK2, prokineticin 2; PROKR2, prokineticin receptor 2; SF1, steroidogenic factor 1 (also FTZF1 or NR5A1); DAX1, dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on X chromosome 1 (also NROB1); HESX1, homeobox expressed in ES cells 1, homeodomain transcription factor; LHX3, LIM/homeobox protein; PROP1, prophet of Pit1; GPR54, G-protein receptor 54; LEP, leptin; LEPR, leptin receptor; GNRHR, gonadotropin-releasing hormone (GnRH) receptor; LHB, luteinizing hormone beta (LH-β); FSHB, follicle-stimulating hormone beta (FSH-β). The hypothalamic-pituitary-gonadal (HPG) axis is depicted: Hypothalamic GnRH pulses stimulate pituitary LH and FSH pulses that act on the gonads to stimulate gametogenesis and sex steroids (T, testosterone; Ez, estradiol), which feed back to regulate the hypothalamus and pituitary. The influence of energy balance is shown by the effect of leptin and ghrelin on the hypothalamus and pituitary.
ADRENARCHE
The adrenal cortex in humans secretes three classes of steroid hormones: glucocorticoids, mineralocorticoids, and androgen precursors. The androgen precursors are mainly dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEAS), and androstenedione. These steroids do not bind to androgen receptors or act as androgens per se. Androgenic potency is derived from conversion in peripheral tissues (e.g., skin, hair follicle, and liver) to the potent androgens testosterone and DHT.
During development, the human adrenal consists of fetal and adult zones.28 Patients with X-linked adrenal hypoplasia congenita (AHC), which is caused by mutations in the orphan nuclear receptor DAX1, develop adrenal insufficiency because the adult zone does not form.29 The fetal zone secretes large amounts of DHEAS because of relatively low expression of 3β-hydroxysteroid dehydrogenase (3β-HSD). The fetal zone normally regresses in early infancy, and the adult zone persists and forms the characteristic zona reticularis, zona fasciculata, and zona glomerulosa. In adolescents and adults, DHEAS is synthesized mainly by the zona reticularis.30
Adrenarche is defined as the time when adrenal androgens, principally DHEA, DHEAS, and androstenedione, increase in a process that is largely independent of sexual maturation.31 Adrenarche is associated with increased growth of pubic and axillary hair and occurs before increases in gonadal sex-steroid secretion.32 Premature adrenarche, which occurs more commonly in girls than boys, is associated with precocious development of axillary and pubic hair and is caused by early increases in adrenal DHEAS production. The usual age of adrenarche onset is about 6 years. Studies of 17-ketosteroid excretion published in the early 1940s showed that at about 6 years of age, 17-ketosteroid excretion begins to increase and rises steadily thereafter to adult values by about ages 20 to 21.33,34 These observations have been confirmed with serum steroid measurements (Fig. 120-4).
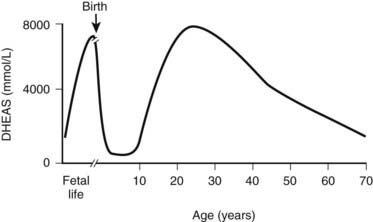
FIGURE 120-4. Variation in circulating dehydroepiandrosterone sulfate (DHEAS) concentration throughout human life. DHEAS concentrations reach a peak at term; following birth, there is a rapid decline. At about 6 years of age, DHEAS concentrations rise again (adrenarche) and reach a peak during early adulthood, declining thereafter in a process often called adrenopause.
(From Auchus RJ, Rainey WE: Adrenarche: physiology, biochemistry, and human disease, Clin Endocrinol (Oxf) 60:288–296, 2004.)
The factors that regulate adrenarche remain enigmatic. Adrenocorticotropic hormone (ACTH) is necessary and is an important modulator of adrenal androgen as well as glucocorticoid secretion. Absence of adrenarche is noted in patients with ACTH-receptor mutations.35 However, the relative potency of ACTH to control adrenal androgen versus cortisol secretion is much less, by a factor of about 1000.36 Furthermore, ACTH levels do not rise at the time of adrenarche, indicating that other factors are involved. Two of the purported factors are cortisol-releasing hormone (CRH) and prolactin (PRL). Exogenous CRH leads to elevated levels of DHEAS in humans. Prolactin receptors are expressed in the adrenal cortex, and PRL stimulates CRH release in the rat hypothalamus.35 POUF1 deficiency includes absence of growth hormone (GH), thyroid stimulating hormone (TSH), and PRL. Patients with POUF1 deficiency demonstrate absent or delayed adrenarche, perhaps because of PRL deficiency.35 In addition to ACTH, CRH, and PRL, several other hormones may modify adrenal androgen secretion. These hormones include estrogens, GH, gonadotropins, lipotropin, and a postulated pituitary hormone called cortical androgen-stimulating hormone.36 Under selected circumstances, each of these hormones may affect adrenal androgen secretion, but none appears to be a major modulator. Of note, estrogen treatment of prepubertal and pre-adrenarchal girls with gonadal dysgenesis commonly leads to the development of axillary and pubic hair, as well as inducing estrogenic effects. However, serum DHEA and DHEAS do not change significantly in such children.37,38 Both estrone and estradiol are inhibitors of 3β-hydroxysteroid dehydrogenase. Thus estrogens, directly or indirectly, can modify adrenal steroidogenesis, in turn affecting the onset of adrenarche.
Although the factors regulating adrenarche remain uncertain, it appears that changes in the constitution of adrenal cells, and their specific enzymatic pathways, account primarily for variations in adrenal androgen production. The activities of CYP11A, CYP17, and SULT2A1, all of which are abundant in the zona reticularis, direct steroidogenesis towards production of DHEAS.32 In addition, DHEAS production is increased by expression of cofactor proteins such as cytochrome b5, which enhance the 17,20-lyase activity of CYP17.
Abnormalities in the timing and intensity of adrenarche may be linked to birth weight, insulin resistance, and polycystic ovarian syndrome (PCOS).39,40 The risk of premature adrenarche is increased in small-for-gestational-age infants.41 Subsequently, girls with premature adrenarche tend to have higher rates of metabolic disease and to be larger in height and weight.42 The risk of PCOS is increased as well, particularly in those children with exaggerated adrenarche.43
CHANGES IN BONE MINERAL DENSITY DURING SEXUAL MATURATION
In a study of 403 white Dutch children, dual-energy x-ray absorptiometry was used to estimate BMD and then correlated with data on pubertal stage, fat mass, and lean tissue mass.44 The percentage of body fat was higher in girls than boys at all ages and was increased in girls in consecutive pubertal stages. In contrast, body fat was lower in stage 4 than in stage 3 in boys. Tanner stage was positively correlated with BMD in both boys and girls and also with percent body fat in girls. In a separate study of 295 girls and 205 boys, spinal BMD correlated positively in both sexes with calcium intake, pubertal stage, and physical activity.45 The major determinant of BMD was Tanner stage in girls and body weight in boys. In a longitudinal study of 68 males and 72 females, over 35% of total body bone mineral and 27% of bone mineral at the femoral neck was found to be laid down in a 4-year adolescent period during peak linear growth velocity.46 This corresponds to as much bone mineral as most adults lose during their remaining life. In a prospective study of peak height velocity and peak bone mineral content (BMC) velocity, peak BMC velocity peaked 1.2 years after peak height velocity in boys and 1.6 years after peak height velocity in girls.47 Within 3 years before or after peak BMC velocity, boys consistently had higher BMC than girls, and this difference increased steadily throughout puberty. Calcium accretion into bone was estimated to be about 500 mg/day at time of peak BMC velocity.
In summary, BMD increases in parallel with pubertal stage in both sexes. The bone mineral accretion rate is very high during these years, and attainment of optimal peak bone density demands a high calcium intake of over 1 g daily during adolescence. Studies in humans and in animal models have suggested that estrogen, formed from aromatization of testosterone in boys and directly secreted from the ovary in girls, is a key mediator of this increasing bone density.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


