FIGURE 119-1. The three stages of normal sexual differentiation. 1, Chromosomal sex is determined by the sex chromosome (X or Y) present in the spermatozoon at fertilization. 2, Gonadal sex differentiation does not occur before the sixth week, when the indifferent gonadal ridge takes the testicular or the ovarian pathway, according to the gene expression pattern. 3, Sex of the internal and external genitalia depends on the secretion and action of two testicular hormones: testosterone and anti-Müllerian hormone (AMH). Male genital differentiation occurs when androgens (testosterone and dihydrotestosterone) and AMH are present and active, whereas female differentiation takes place in their absence.
Nomenclature and definitions of the various disorders of gonadal and genital development have been discussed recently by experts who produced a consensus statement on the management of intersex disorders.3,4 The authors agree that the use of terms such as intersex, hermaphroditism, pseudohermaphroditism, and sex reversion is controversial and could be perceived as pejorative by patients. Therefore, they have proposed the use of “disorders of sex development (DSD)” to define congenital conditions in which chromosomal, gonadal, and genital sex are not coincident. In this chapter, we will address especially those conditions in which the development of the gonads and/or genitalia is atypical. Although they do not represent a differential diagnosis of any intersex disorder, conditions limited to atypical chromosomal sex, such as Klinefelter and Turner syndromes and other sex chromosome aneuploidies, will be discussed in this chapter.
Classification of DSD is complex. Based on recognition of the underlying anomaly in the process of sex organ development (Table 119-1), cases of DSD may be divided into (1) malformative DSD, wherein abnormal morphogenesis of the genital primordia occurs in early embryonic life; (2) dysgenetic DSD, due to abnormal gonadal differentiation; and (3) non-dysgenetic DSD, in which abnormal sex hormone–dependent genital differentiation occurs in the presence of non-dysgenetic gonads. Abnormal morphogenesis of the genital primordia may be due to chromosomal, genetic, or environmental causes that impair the normal embryologic processes underlying the initial formation of the indifferent urogenital ridge, urogenital sinus, and primordium of the external genitalia. Abnormal gonadal differentiation, or gonadal dysgenesis, is mainly dependent on sex chromosome defects or mutations in genes involved in testis or ovary differentiation. Finally, abnormal sex hormone–dependent genital differentiation includes two categories: (1) 46,XY individuals with differentiated testes but ambiguous or female genitalia, resulting mainly from abnormal male hormone synthesis or action (previously named male pseudohermaphrodites), and (2) 46,XX individuals with normal ovaries and female internal genitalia but ambiguous or male external genitalia, resulting mainly from exposure to abnormally high levels of androgens in utero at the critical time of sex differentiation (previously named female pseudohermaphrodites). DSD may occur as isolated abnormalities or as part of polymalformative syndromes.
Table 119-1. Classification of Disorders of Sex Development (DSD)
It is very important to recognize DSD as early as possible in life and to not delay the etiologic diagnosis. The diagnosis should be as precise as possible for instituting substitutive treatment in case of an enzymatic defect in steroid biosynthesis but also primarily for helping the choice of the most appropriate gender for rearing. Investigations should anticipate the chances of the best somatic and psychological development in the sex assigned. Because DSD may be caused by genetic abnormalities as well as by environmental factors, genetic counseling is possible only when the cause and the mode of transmission are known.
ABNORMAL MORPHOGENESIS OF THE UROGENITAL PRIMORDIA (MALFORMATIVE DSD)
During the fourth and fifth weeks of embryonic life, two mesoderm derivatives, the mesonephros and the coelomic epithelium, originate the anlagen of the gonads and of the internal—Wolffian and Müllerian—reproductive ducts (Fig. 119-2), together with those of other nonreproductive organs like the adrenal cortex and the renal excretory system. The distal part of the hind gut, or cloaca, is shut off by the cloacal membrane. During the fourth week, the urorectal septum separates the urogenital sinus, ventrally, from the rectum. The cloacal membrane is also divided into the urogenital membrane and the anal membrane. The urogenital sinus gives rise to the bladder in both sexes, and to the whole urethra and the lower part of the vagina in the female, or to the prostate and the prostatic and membranous urethra in the male. The urogenital membrane is surrounded by the genital folds (or labioscrotal swellings). Ventrally, the genital tubercle emerges as a medial outgrowth between the genital folds (Fig. 119-3). After the corpora cavernosa and glans have differentiated, the ventral surface of the genital tubercle is depressed by a deep furrow, the urethral groove giving rise to the spongy (or penile) urethra. The external genitalia remain undifferentiated up to approximately 9 weeks. The urogenital membrane folds and gives rise to the labia minora in the female or the penile urethra in the male, the labioscrotal swellings differentiate into the labia majora or the scrotum, and the genital tubercle gives rise to the glans clitoridis or glans penis. The underlying morphogenetic processes begin to be unraveled (for a detailed description, see reference 5 and Chapter 118).
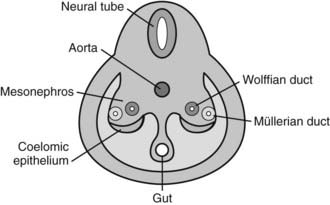
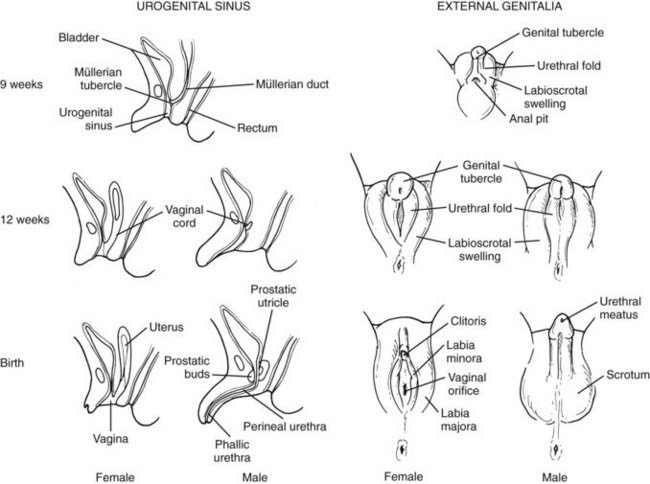
FIGURE 119-3. Sex differentiation of urogenital sinus (left) and external genitalia (right).
(Modified from Rey R, Josso N. Sexual differentiation. In De Groot LJ (ed): Endotext.org, Updated April 10, 2007. http://www.endotext.org/pediatrics/pediatrics7/pediatricsframe7.htm.
Defective Morphogenesis of the Urogenital Sinus and of the Primordium of the External Genitalia, including Isolated Hypospadias, Aphallia, and Cloacal Malformations
It is easily conceivable that inadequate morphogenesis of the genital primordia during early embryonic life independently from hormone action will result in abnormalities of genital development. In some cases, for instance in cloacal malformations, perineal anatomy may be so affected that the sex of the external genitalia cannot be identified.6 Although urologic and hind gut anomalies are the major problems to be solved in these cases, sex assignment could also be a serious issue. Factors that influence gender assignment do not differ from those discussed in section “Patients with Polymalformative Syndromes.” Karyotype and hormone laboratory evaluations are useful for establishing gonadal sex and function. The cause remains unknown in most cases; recently a chromosomal aberration has been identified as a possible cause.7
Non–endocrine-related DSD should be considered when there is inconsistency between the development of the different elements of the genitalia (Table 119-2). An example is isolated congenital aphallia,8 in which the rest of the genitalia have a normal configuration. Many factors are known to drive the early morphogenesis of the external genital primordia.9 In most cases, endocrine-unrelated malformations of the genitalia are associated with other somatic dysmorphic features. Micropenis or aphallia has been observed in Robinow syndrome due to ROR2 mutations (OMIM 180700); micropenis and hypospadias, in Pallister-Hall syndrome due to GLI3 mutations (OMIM 146510); and hypospadias, in the X-linked Opitz G/BBB syndrome, which can be caused by mutations in the MID1 gene (OMIM 300000), as well as in the hand-foot-genital syndrome associated with HOXA13 defects (OMIM 140000), in Rieger syndrome type 1 caused by mutations in the homeobox transcription factor gene PITX2 (OMIM 180500), and in velocardiofacial syndrome associated with a hemizygous deletion of chromosome 22q11.2, resulting in haploinsufficiency of the TBX1 gene (OMIM 192430). Isolated hypospadias (i.e., with no other signs of hypovirilization, or in the context of polymalformative syndromes) has a low risk of being due to anomalies of gonadal hormone action.10
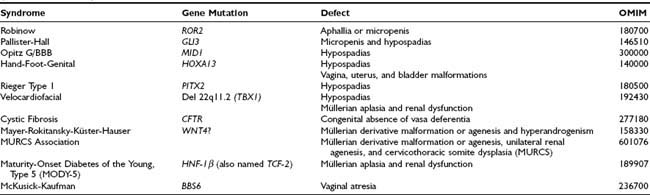
Defective Morphogenesis of the Gonadal Ducts, Including Rokitansky Syndrome and Absence of the Vasa Deferentia
Malformations of the vagina, uterus, and bladder are present in females with hand-foot-genital syndrome due to HOXA13 mutations (OMIM 140000). Mayer-Rokitansky-Küster-Hauser syndrome (MRKH) is a heterogeneous disorder characterized by uterovaginal atresia in 46,XX females (see Table 119-2). Abnormalities of the genital tract may range from upper vaginal atresia to total Müllerian agenesis associated with urinary tract abnormalities,11 and even cervicothoracic somite dysplasia (MURCS association, OMIM 601076). Although initially hypothesized to be due to abnormal activation of AMH expression or AMH receptor signaling in the female fetus, no mutations have been found in these genes.11,12 Mutations in the WNT4 gene have been identified in a small subset of MRKH patients with signs of hyperandrogenism and female range AMH.13–15 However, even if WNT4 is clearly involved in early Müllerian duct embryogenesis,16 it does not seem to be the main factor responsible for MRKH syndrome.15,17 Müllerian aplasia has also been described in patients with renal dysfunction and maturity-onset diabetes of the young type 5 (MODY-5) due to a mutation in HNF-1β (also named TCF-2) gene18 and in two patients with velocardiofacial syndrome due to a deletion of chromosome 22q11.2.19 Vaginal atresia has been described in McKusick-Kaufman syndrome (OMIM 236700), probably caused by mutations in the BBS6 gene.
Congenital bilateral absence of the vas deferens is responsible for 1% to 2% of cases of male infertility and is present in 95% of patients affected with cystic fibrosis (OMIM 277180),20 a bronchial and pancreatic disease due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). Whether efferent duct maldevelopment is a primary defect of cystic fibrosis or a secondary degenerative change resulting from obstruction by mucus is not known at the present time. Isolated absence of vas deferens in otherwise healthy men is often associated with the presence of a single CFTR allele mutation.20
ABNORMAL GONADAL DIFFERENTIATION (DYSGENETIC DSD)
As established by Jost,2 dual hormonal secretion by the testis in early fetal life is a determinant for the differentiation of the internal and external genitalia. Furthermore, the processes of genital differentiation in most cases are subjected to tight regulation whereby timing is critical. For instance, in the male, AMH must be expressed before the end of the eighth week, when Müllerian ducts begin to lose their responsiveness.21 Therefore, any disorder affecting the normal process and/or timing of testicular development will result in a DSD. Conversely, because female differentiation of the genitalia occurs in the absence of hormone action, defective ovarian development has no effect on fetal sexual development. This biological phenomenon has two practical consequences: DSD have been best characterized in XY patients with defective testicular development, and, consequently, the normal process of testicular differentiation has been studied more extensively than that of ovarian differentiation (see Chapter 118).
Gonadal dysgenesis may arise from early defects of gonadal primordium formation, or from specific defects in differentiation of the testis or the ovary. However, the clinical outcomes in terms of gonadal histophysiology and genital anatomy are similar, and finally depend on the amount of functional testicular tissue that has developed. Gonadal dysgenesis represents the most conspicuous form of fetal-onset primary hypogonadism. Its severity may range from complete gonadal dysgenesis affecting all gonad-specific cell lineages—Leydig/theca, Sertoli/granulosa, and germ cells—to extremely mild dysgenesis. It should also be considered that the condition may affect the right and left gonads equally, or may be asymmetrical. Finally, gonadal chimeras consisting of both ovarian and testicular tissue may form; these are called ovotestes. All these defects of gonadal formation, differentiation, or maintenance are part of a single wide spectrum of disorders.
We will address first the clinical presentations of gonadal dysgenesis, and then we will discuss the mechanisms underlying defective formation of the fetal gonads.
Anatomic and Clinical Aspects of Dysgenetic DSD
Complete Gonadal Dysgenesis
Irrespective of its origin, gonadal dysgenesis can be complete, partial, or extremely mild. Patients with streak gonads and normal female gonadal ducts and external genitalia represent by definition the complete form of the so-called pure or complete gonadal dysgenesis. The streak gonads do not secrete sex steroids or AMH.
Complete Gonadal Dysgenesis in Patients With a Y Chromosome: Swyer Syndrome
In 1955, Swyer22 described two adult XY females presenting with tall stature and eunuchoidal proportions, female internal and external genitalia (one had mildly enlarged clitoris), little or no mammary development, and primary amenorrhea. These two cases represent almost complete pure gonadal dysgenesis. In the complete form, primary amenorrhea with sexual infantilism is the most frequent presenting feature in a child reared as a female who has a normal or tall stature and no stigmata of Turner syndrome. Such a child has a 46,XY karyotype and no evidence of mosaicism. Serum testosterone and AMH are undetectable,23 and gonadotropin levels are elevated even during childhood (Table 119-3).24 A uterus may be demonstrated on ultrasonography. At laparotomy, two streak gonads with an ovary-like stroma but absent ovarian follicles are found. An increased risk exists for the development of gonadal tumors, gonadoblastoma, and dysgerminoma in XY patients.25
Table 119-3. Differential Diagnoses in 46,XY Disorders of Sex Development (DSD) With Normal Adrenal Function
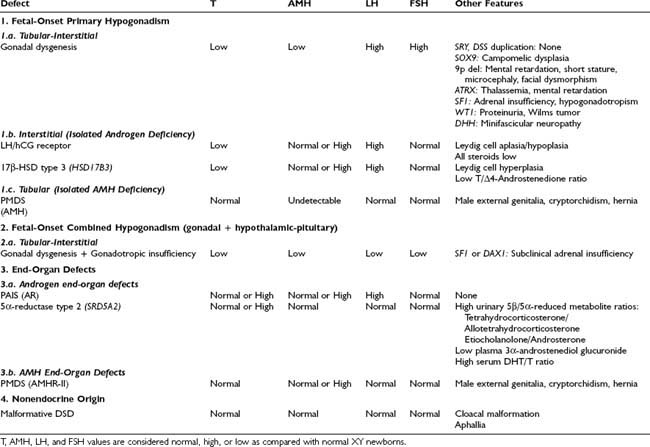
The 46,XY gonadal dysgenesis syndrome can be sporadic or familial. Its origin is heterogeneous and may be genetic or environmental. Familial reported cases are compatible with X-linked recessive transmission or sex-limited dominant autosomal transmission.26,27
46,XX Complete Gonadal Dysgenesis
Patients with pure gonadal dysgenesis and 46,XX karyotype have a normal stature, sexual infantilism, and bilateral streak gonads. A marked heterogeneity, both genetically and clinically, has been noted in the expression of the disease, even in the same kindred. The molecular basis of the condition is still not well understood. The frequency of consanguinity in affected families points to an autosomal gene necessary for normal ovarian development and function. Alternatively, the condition may result from an X-linked gene mutation or deletion in sporadic cases. Adult height is lower in 46,XX than in 46,XY gonadal dysgenesis, which suggests the existence of a Y-specific growth gene that promotes statural growth independently of gonadal steroids.28 The main clinical features are the lack of stigmata of Turner syndrome. Presenting signs at the age of puberty include lack of breast development, primary amenorrhea, and elevated gonadotropins.
45,X Complete Gonadal Dysgenesis: Turner Syndrome
It was described by Turner in 1938 as a distinct entity associating sexual infantilism, webbed neck, and cubitus valgus.29 As early as 1930, Ullrich had in fact described the same physical characteristics in female patients.30 In subsequent years, gonadal dysgenesis was recognized as part of Ullrich-Turner syndrome (called exclusively Turner syndrome in the Anglo-Saxon literature). Definitive evidence for X-chromosomal monosomy was provided in 1959.31 Turner syndrome occurs in approximately 1 in 2500 live female births. The number of unborn affected fetuses is much larger, however, because approximately 98% of pregnancies with Turner syndrome abort spontaneously for reasons that are not yet understood.
The syndrome spans a wide spectrum of clinical presentations.32,33 The main characteristics are abnormal physical features, gonadal dysgenesis, and short stature. Children with Turner syndrome may also have the following physical findings (Fig. 119-4): short, thick, and webbed neck; dysmorphic face (fish-like mouth, hypertelorism, epicanthus, ptosis of the eyelids, prominent ears, high-arched palate, micrognathia) and low posterior hairline; congenital lymphedema (puffy hands and feet at birth); broad child-like chest; hypoplastic or inverted nipples; cubitus valgus; short fourth metacarpals; multiple pigmented nevi; abnormal fingernails (turned out at the end); intestinal telangiectasia; recurrent otitis media; and a tendency to keloid formation. Turner patients also show a prevalence of celiac disease in childhood or adolescence. Cardiovascular defects, especially coarctation of the aorta and nonstenotic bicuspid aorta, are common. A routine echocardiogram thus is indicated in all 45,X patients. Kidney anomalies occur in one third to one half of girls with Turner syndrome, with monosomic patients at greater risk. The most common anomaly is a horseshoe kidney. The prevalence of mental retardation appears no greater than in the general population. Many patients frequently exhibit gross and fine motor dysfunction. Bone age is delayed and mineral bone density may be affected.
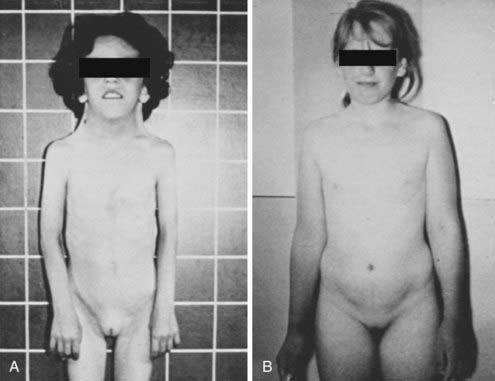
FIGURE 119-4. Phenotypic spectrum in patients with 45,X Turner syndrome. A, The girl has typical morphologic anomalies, in particular, a pronounced webbed neck. B, The girl has very few morphologic anomalies. Both patients had short stature.
The newborn with Turner syndrome is smaller than average both in length and in weight.34 Usually, postnatal growth lies in the normal range for the first 2 to 3 years but then decreases. Height further deviates from the norm with age. Haploinsufficiency of SHOX, mapping to Xq22, is clearly involved in the short stature phenotype of Turner syndrome patients.35 However, the deficit in height is due mainly to the lack of pubertal growth spurt. The ultimate mean height is below the third percentile (ranging from 139 to 147 cm). Family height plays a role in determining the ultimate height in girls with Turner syndrome,36,37 and significant differences in final height have been noted in Turner patients from different ethnic groups. Growth charts adapted to Turner’s spontaneous growth have been established in various populations.34 Body proportions are altered (increased upper/lower segment ratio) in most cases, and this is not improved by growth hormone (GH) therapy. Furthermore, treatment seems to aggravate the disproportionate growth of feet.38 An increase in subcutaneous adipose tissue is seen with age and frequent obesity.39
Gonadotropin levels are found above the normal range throughout childhood in girls with Turner syndrome.24 Thyroid hormones are usually normal, although subclinical thyroid dysfunction may be present. The incidence of chronic lymphocytic thyroiditis, diabetes mellitus, rheumatoid arthritis, and inflammatory bowel disease is increased.
Partial Gonadal Dysgenesis
Partial Gonadal Dysgenesis in Patients With a Y Chromosome (Dysgenetic Male Pseudohermaphroditism)
The phenotypic differences observed in partial gonadal dysgenesis in patients with a Y chromosome (previously named dysgenetic male pseudohermaphroditism) depend on the extent of testicular differentiation. Patients with partial gonadal dysgenesis may have a mixture of Wolffian and Müllerian duct derivatives and some functional capacity to produce testosterone and AMH. A continuum of phenotypes ranging from female genitalia with clitoromegaly to male genitalia with mild hypospadias and cryptorchidism has been described. In the most severe cases of partial gonadal dysgenesis, gonadal tissue may contain areas of poorly differentiated seminiferous tubules in combination with areas of ovary-like stroma. These streak testes should be distinguished from ovotestes, in which ovarian follicles are present.40 All dysgenetic gonads have an increased risk for gonadal tumors.3,4 Spontaneous breast development may provide the first evidence of an estrogen-secreting gonadal tumor. In milder forms, testes have distinct seminiferous tubules; however, the gonads are small, the albuginea is thin and loosely organized, and the seminiferous tubules are separated by wide intertubular spaces, frequently forming a network that resembles the first stages of sex cord formation in the developing testis and contains scarce germ cells (Fig. 119-5A through 119-5C).40 Ring tubules, calcified psamomma bodies, or a persistent coelomic epithelium can also be observed. A frequent surgical finding is the dissociation between testis and epididymis, joined only by a thin connective membrane. Serum testosterone, AMH, and inhibin B are intermediate between the male and female ranges, and gonadotropins are elevated, in correlation with the degree of dysgenesis.23,41,42

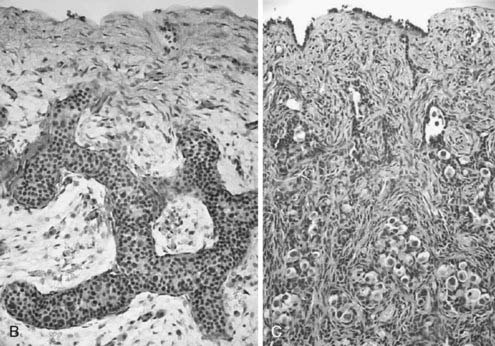
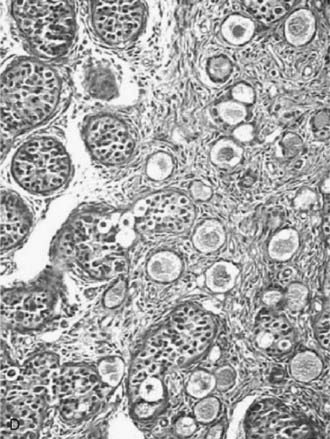
FIGURE 119-5. A, Normal testis of an 11-week-old fetus: anastomosing seminiferous cords, positively stained for anti-Müllerian hormone (AMH), form a typical network. B, Dysgenetic testis with abnormal seminiferous cords forming an anastomosing network. C, Streak gonad with abnormal germ cells. D, Ovotestis, showing seminiferous cords and follicles with oocytes.
(Modified with permission from Chemes H, et al: Early manifestations of testicular dysgenesis in children: pathological phenotypes, karyotype correlations and precursor stages of tumour development, APMIS 111:12–24, 2003.
45,X Partial Gonadal Dysgenesis: Turner Syndrome Variants
Partial X chromosome losses may also occur, leading to isochromosomes made from two parts of the long i(X)q or two parts of the short chromosome segment i(Xp), or from major deletions (denoted “del” or “−”). Minor deletions, or the loss of small X fragments, can be visualized only through banding techniques or with the use of DNA probes. Finally, an X chromosome may fail to develop in the usual rod form, but instead may close to form a ring; the karyotype then is 46,X,r(X). The normal X chromosome comes from the mother in most patients with a 45,X karyotype, but only in about half of those with a isochromosomes or ring chromosomes.43 The percentage of 45,X chromosomes versus mosaics or isochromosomes varies somewhat among populations.
Mosaic forms of Turner syndrome usually are seen in female adolescents with primary amenorrhea and in young women with premature ovarian failure. It is estimated, however, that 3% to 8% of 45,X karyotype patients and 12% to 21% of females with sex chromosome mosaicism may have normal pubertal development and spontaneous menstrual periods, but their final height is not significantly greater.44 Spontaneous pregnancy is an exceptional event that occurs in approximately 2% of cases and more often in patients with structural anomalies of the X chromosomes in which the Xq13-q26 region, containing the genes that are thought to control ovarian function, is spared, or in patients with a mosaic karyotype containing a 46,XX cell line (45,X and 45,X/46,XX), which preserves ovarian development.
Asymmetrical Gonadal Differentiation, Including Ovotesticular DSD (True Hermaphroditism)
The degree of gonadal dysgenesis may be different in the two gonads. A great variety of possibilities exist: a dysgenetic testis or ovary on one side and a streak (Fig. 119-5D) contralaterally; a dysgenetic testis on one side and a dysgenetic ovary on the other; an ovotestis on one side and a streak or a dysgenetic testis or ovary contralaterally; and so forth. All these are combinations of asymmetrical gonadal development. The resulting phenotype depends on the amount of functional testicular tissue that exists on each side of the body.
As suggested by Jost’s pioneering experiments,2 the action of fetal testicular hormones is mainly local. This can be observed clearly in asymmetrical gonadal differentiation,45 also called mixed gonadal dysgenesis,46 characterized by the existence of a mildly dysgenetic testis on one side and a severely dysgenetic or streak gonad contralaterally. Patients usually present with male gonadal ducts and a palpable gonad in a hemi-scrotum on one side (Fig. 119-6) and a hemi-uterus and an empty labioscrotal fold on the other side. The size of the phallus and the position of the urethral meatus are variable. The most commonly found karyotype in peripheral blood is 45,X/46,XY, but karyotype may range from normal 46,XY to different mosaicisms. When studied in gonadal or streak tissue, a 46,XY karyotype is more prevalent in the normal gonad, and abnormal karyotypes are found in the streak.40 As in other cases of dysgenetic DSD, serum testosterone, AMH, and gonadotropins correlate with the severity of the condition.23,42
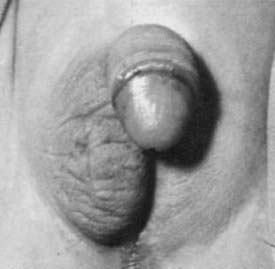
FIGURE 119-6. Patient with asymmetrical gonadal differentiation (also known as mixed gonadal dysgenesis). Note the asymmetrical development of the external genitalia.
Ovotesticular DSD, previously known as true hermaphroditism, refers to individuals who have both ovarian and testicular tissues in the same gonad (see Fig. 119-5D) or in contralateral gonads. Ovotesticular DSD is relatively rare, except in blacks from Southern Africa, in whom it is the most common intersex condition.47 The bisexual gonad contains testicular tissue with distinct tubules, and the ovarian tissue has follicles. The ovarian tissue must contain oocytes for the diagnosis, and the presence of only ovarian stroma is not an adequate criterion.40 As in other cases of dysgenetic DSD, the degree of genital ambiguity depends on the functional mass of testicular tissue present in these patients. The ovotestis is the most frequent gonad found (59%), and two components are arranged end to end. The ovarian tissue is usually normal, contrasting with dysgenetic testicular tissue lacking spermatogonia. A vas deferens may be palpable. The gonadal descent occurs more frequently on the right side. Internal genital differentiation is variable and reflects gonadal endocrine capabilities. It is often asymmetrical. According to the nature and the location of the gonadal tissue, a classification has been proposed: (1) bilateral with two ovotestes; (2) lateral with testis on one side, ovary on the other; and (3) unilateral with a normal gonad on one side and an ovotestis on the other side, the most frequent condition. The descended gonad usually contains some testicular tissue.48
Normal female phenotype and almost normal male appearance with penile hypospadias or small penis have been reported. In some patients who are reared as females, virilization may cause hirsutism and clitoromegaly. In others, normal ovarian function and fertility may even occur. In phenotypic males, gynecomastia and urethral bleeding may occur at pubertal age. In adulthood, testosterone levels may be sufficient and patients may be fertile; however hypoandrogenism and infertility are more common.
The most frequent karyotype is 46,XX, followed by various forms of mosaicism (46,XX/46,XY, 45,X/46,XX/47,XYY, etc.).48 Hormonal laboratory studies usually correlate with the severity of the gonadal dysgenesis: when the testicular tissue is abundant and normal, gonadotropins, testosterone, and AMH may be in the normal male range.23,42 Conversely, when ovarian tissue is predominant, estradiol may be in the normal female range. When the gonads are more dysgenetic, FSH is usually elevated, and gonadal steroids and peptides are low. The risk for tumor development remains unclear.3,4
Mild Gonadal Dysgenesis
46,XY Mild Gonadal Dysgenesis
In the mildest forms, testes are almost normal, except for a reduced number of germ cells. Patients have normal male genitalia and hormonal levels within the expected range for sex and age. In some cases, gonadal dysgenesis is suspected by testicular-epididymal dissociation observed during surgery for cryptorchidism and is confirmed by histologic examination. In other cases, the first manifestation is observed in adults who present with infertility or testicular tumor.49
46,XX Males
This condition, previously called XX sex reversal, is characterized by testicular development in subjects who have two X chromosomes but lack a normal Y chromosome. Approximately 1 in 20,000 to 30,000 males have a 46,XX karyotype.50 Most cases are sporadic, and a form of X-Y paternal interchange is suggested when part of the short arm of the Y chromosome carrying SRY is transferred to one of the two X chromosomes.51 The high sequence identity and identical orientation of PRKY to its homologous gene on the X chromosome, PRKX, seem to explain the high frequency of abnormal pairing and subsequent ectopic recombination outside the pseudoautosomal region, leading to XX males and XY females (Fig. 119-7).52 SRY(+) XX males usually have a normal male phenotype with no particular feature during childhood, and the condition is often diagnosed at puberty or adulthood owing to small testes, gynecomastia, and sterility. Some degree of testosterone deficiency with elevated plasma gonadotropins has been reported. Owing to the presence of two X chromosomes and the lack of Y chromosome genes involved in spermatogenesis, germ cells do not go through meiosis, which explains the small testis size and azoospermia, as is seen in Klinefelter syndrome. However, XX males show shorter stature than Klinefelter patients.
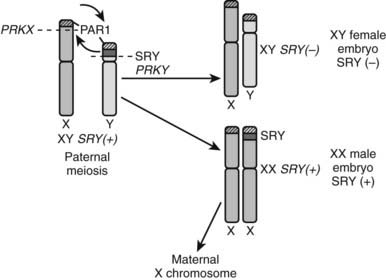
FIGURE 119-7. Origin of SRY-negative XY females and SRY-positive XX males. During meiosis in the paternal germ cells, an abnormal translocation occurs of SRY from the Y chromosome to the X chromosome (e.g., at the level of the PRKX and PRKY genes). If the spermatozoon carrying an X chromosome with SRY fertilizes an oocyte, an SRY-positive XX embryo results. Conversely, if the spermatozoon carrying the SRY-negative Y chromome fertilizes an oocyte, an SRY-negative XY embryo results.
A small proportion of XX males lacking SRY sequences have been described. They differ from 46,XX SRY(+) patients by the occurrence of hypospadias, micropenis, and/or cryptorchidism.53,54 These SRY(−) XX males with abnormal external genitalia clearly have more severely dysgenetic gonads and closely resemble XX ovotesticular DSD.55 Because of insufficient sampling of the gonadal tissue, the diagnosis of ovotesticular DSD may be missed.
Mild Gonadal Dysgenesis in Sex Chromosome Aneuploidies: XXY, XYY, XXX, and Other Aneuploidies
Klinefelter Syndrome
Klinefelter syndrome, the most common cause of male hypogonadism, with a prevalence of approximately 1.5 in 1000 males, was first described in 1942 as the association of eunuchoidism, gynecomastia, small testes, azoospermia, and elevated FSH excretion.56 Several groups subsequently reported that a high proportion of patients with this syndrome were chromatin positive, a finding explained in 1959 when the XXY constitution was reported.57 Patients with the 47,XXY genotype have classic Klinefelter syndrome. Over the past three decades, the clinical spectrum of the syndrome has expanded to include patients with pure cell lines or mosaicisms with two or more X chromosomes and a Y chromosome.58
Gonadal dysgenesis primarily affects germ cells beginning in early fetal life59 but does not impair Leydig and Sertoli cell function until midpuberty.60 Therefore, internal and external genitalia are normally virilized in Klinefelter patients.
Generally, at birth, no particular stigmata occur in Klinefelter syndrome. During childhood, no specific signs or symptoms are noted, except for bilateral cryptorchidism, learning disabilities, or behavior disorders, which are frequent.61,62 Klinefelter boys are long-legged, most probably as the result of an overdose of SHOX gene expression.63 The pathogenesis of personality disturbances, intellectual deficits, and associated medical disorders remains unexplained.
Pubertal development usually occurs spontaneously. In all cases, the predominant features are the small size of the testes (100%), which do not increase normally at puberty, and pubertal gynecomastia (60%). At puberty, as the gonadotropins increase, the seminiferous tubules do not enlarge, but rather undergo fibrosis and hyalinization, which results in small firm testes. Obliteration of the seminiferous tubules results in azoospermia. The marked reduction of tubular volume accounts for the apparent hyperplasia of the interstitial tissue. Leydig cells are present in clumps, and although their total mass is normal, they are functionally abnormal, resulting in a reduced testosterone production rate with compensatory luteinizing hormone (LH) hypersecretion. Because of the high LH levels, estradiol secretion is stimulated, with an increased estradiol/testosterone ratio, which is believed to account for the development of gynecomastia.58,63
47,XYY males and 47,XXX females
Although the prevalence of these conditions is approximately 1 in 1000 live births,62 they are rarely diagnosed. Most cases are oligo- or asymptomatic. Gonadal development and function is normal in the vast majority of cases, except for a few XXX women who have been reported to develop premature ovarian failure.64 Children are long-legged, with accelerated growth from infancy resulting in increased height at pubertal onset and might show behavior disturbances.62,63,65
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


