FIGURE 31-1. Schematic outline of the development of the mammalian pancreas. 1, Dorsal and ventral buds arise from the duodenal part of the primitive gut endoderm (blackened). 2, The dominant ventral bud, following the bile duct, turns and approaches the dorsal bud. 3, The two buds fuse together. 4, The ventral bud becomes part of the head of the pancreas (the PP-rich duodenal lobe), whereas the dorsal bud gives rise to the body and tail (the glucagon-rich splenic lobe); the pancreatic juice is drained off by the main and accessory pancreatic ducts. b, bile duct; d, dorsal bud; s, stomach; v, ventral bud.
Similar tissue interactions guide the development of the two ventral buds. In contrast to the dorsal bud that forms in the midline of the epithelium, the ventral buds are derived from the ventrolateral edges of the endodermal sheet. Instructive signals provided by the lateral plate mesenchyme initiate ventral pancreas formation, and interaction with smaller blood vessels further supports organ growth.8 One of these ventral buds degenerates, while the other one fuses with the dorsal bud when stomach and gut rotate to form the mature organ. The regulation of this rotation is not well understood, but perturbations lead to malformations in humans, including annular pancreas, wherein a part of the ventral pancreas is mislocalized and can constrict the adjacent duodenum.9
Mesenchymal-epithelial interactions are required for proper development of mature tissue10,11; however, the epithelial cells start to outgrow the mesenchyme during later stages of development, and few remnants of this tissue remain in the adult pancreas. Epithelial cells branch into the surrounding mesenchyme to form an elaborate duct system that allows transport of exocrine enzymes into the duodenum. Distal cells of the branching epithelium differentiate into acinar cells that are connected to the duct system via centroacinar cells. Acinar cells are organized as small glands that produce a variety of digestive enzymes, including amylases, peptidases, nucleases, and lipases.
In contrast to the continuous acinar duct system, endocrine precursors delaminate from immature ducts. These distinct islet cells produce a variety of hormones, including insulin, glucagon, pancreatic polypeptide, and somatostatin, which are secreted directly into the bloodstream and regulate gastrointestinal function and nutrient storage and utilization. Because of intimate interactions between endocrine cells and blood vessels, islets are highly vascularized. Similarly, hormone secretion is controlled at least in part by the nervous system; the sympathetic, parasympathetic, and sensory neurons that innervate the islets are derived from cells that migrate from the neural crest into the pancreas just as the two buds fuse.12
Early Organ Specification and Bud Formation
TISSUE INTERACTION AND SIGNALING PATHWAYS
With the exception of supporting mesenchyme, blood vessels, and innervating neurons, all mature pancreatic cell types are derived from epithelial cells of endodermal origin.13–15 Several studies have addressed the question of how the pancreas anlage becomes specified within the epithelial sheet that forms the endoderm, and increasing evidence points to a series of tissue interactions as important steps during early stages of organogenesis. During gastrulation, the mesoectodermal portion of the embryo signals to the endodermal sheet to establish a prepattern along the anterior-posterior axis.3
Although the exact signals required for patterning of the fore-midgut area are not completely understood, studies in mice have implicated the fibroblast growth factor (FGF) signaling pathway in this process.16 Before any morphologic signs of pancreas formation are apparent, the midline of the endodermal epithelium comes in close contact with the overlying notochord, a mesodermal structure known to regulate organogenesis and cell differentiation in adjacent structures. The notochord provides numerous secreted signaling proteins that are required for initiation of dorsal pancreas organogenesis. Members of the transforming growth factor-β (TGF-β) and FGF signaling pathways have been implicated in the notochord-mediated induction of pancreas development. A critical aspect of their activity involves repression of the expression of Sonic Hedgehog (Shh), a member of the Hedgehog signaling pathway within the pancreas anlage.17 Ectopic elevation of Hedgehog signaling at the onset of pancreas formation results in pancreas agenesis, indicating that tight regulation of the activity of this pathway is essential for proper organ formation. However, the notochord provides only permissive signals because it cannot induce expression of pancreatic markers in nonpancreatic endoderm.6
In contrast to the singular dorsal pancreas, two distinct pancreatic buds form within the ventrolateral endoderm, a region that is not contacted by the notochord. Tissue recombination experiments in chick and mice demonstrate that similar but distinct signals from the lateral plate mesoderm (LPM) ensure the correct temporal-spatial induction of the ventral pancreas.7,16 Before contact is made with the LPM, no pancreatic markers are detectable in the presumptive ventral pancreas, but LPM or the TGF-β superfamily members activin and morphogenetic proteins (BMPs), as well as retinoic acid (RA), have been shown to induce expression of pancreatic genes in underlying endoderm (see the following section).7 It is important to note that mesenchyme isolated from the pancreatic region carries the potential to induce pancreatic gene expression in anterior endoderm normally fated to develop into stomach and esophagus. Thus, in contrast to the notochord-endoderm interaction that induces dorsal pancreas development in a permissive fashion, the LPM actively instructs uncommitted endoderm to differentiate into pancreatic tissue. Activin, BMP, and RA signals that mimic the pancreas instructive activity of the LPM are potentially useful in designing a cocktail of growth factors designed to regulate differentiation of uncommitted progenitor cells toward a pancreatic fate.
Other studies have shown that interactions between heart mesenchyme and ventral foregut endoderm regulate differentiation of ventral pancreas and liver progenitor cells.18 In an analogy to the notochord-dorsal pancreas bud interactions, the septum transversum and heart mesenchyme produce BMP and FGF signals, respectively, that regulate organ differentiation of the ventral endodermal organs, including liver and pancreas. Because of its close proximity, the concentration of FGF ligands produced by the heart mesenchyme is higher at the area fated to develop into liver and lower at the more distal region that develops into ventral pancreas.18 This is also the case for dorsal pancreatic epithelium, in which low levels of FGF signals have been shown to initiate pancreatic gene expression and block Shh expression, while higher levels of FGF signaling block pancreas induction via increased Hedgehog signaling. Thus, formation of dorsal and ventral pancreatic buds depends on low FGF signals and inhibition of Hedgehog signaling. It is important to note that Hedgehog signaling is active in areas immediately adjacent to the dorsal and ventral buds, thereby establishing a molecular boundary that prevents ectopic expansion of pancreatic tissue beyond its normal borders.
During subsequent stages, the flattened endodermal sheet expands and folds itself to form a tube-like structure. The first morphologic sign of pancreas formation is an epithelial thickening at the dorsal side of the forming gut tube caudal to the stomach anlage, shortly followed by the appearance of two ventral thickenings next to the liver diverticulum. Outgrowth and tissue-specific gene expression continue to depend on cues derived from adjacent tissues. In contrast to earlier stages, mesenchymal signals are now produced from forming blood vessels, the dorsal aorta, and smaller vitelline veins that contact the dorsal and ventral buds, respectively.19 Depletion of blood vessels via explantation in Xenopus or via homozygous recombination in transgenic mice that lack the Flk-1 gene, a receptor for vascular epithelial growth factor (VEGF), impairs the differentiation of the dorsal pancreatic epithelium.19,20 Vice versa, ectopic expression of VEGF leads to hypervascularization and islet hyperplasia in pancreatic tissues, as well as ectopic insulin expression in nonpancreatic stomach epithelium, suggesting that VEGF or molecules secreted by endothelial cells support endocrine cell differentiation. Detailed analysis of the interaction between blood vessels and pancreas buds has revealed physical contact between the aorta and the dorsal bud epithelium, while vitelline veins and ventral bud epithelium are separated by a fine band of mesenchymal cells, although endothelial cells are less critical for ventral bud formation.20
Recent results have provided further evidence for the role of signaling molecules in setting up organ boundaries in the pancreas anlage. Studies in zebrafish suggest that Fgf10 expression is critical for separating pancreatic and liver cells in the area of the hepatopancreatic ductal system. Fgf10 is expressed in the mesenchyme surrounding the developing hepatopancreatic duct and functions to restrain inappropriate differentiation of adjacent pancreatic into liver cells, as well as liver into pancreatic cells.21 Thus, secreted signaling molecules are essential in defining pancreas organ boundaries.
Upon specification of the dorsal and ventral pancreatic anlagen, additional mesenchymal-epithelial interactions are required for later steps of pancreas organogenesis.10,11 Splanchnic mesoderm expands medially and surrounds the pancreatic epithelial buds. Signals from the mesenchyme promote epithelial expansion that results in the formation of a branching structure composed of undifferentiated ductal cells. Based on morphologic criteria, Rutter and colleagues argued that early endodermal cells are “protodifferentiated,” and that mature exocrine and endocrine cells appear only after the secondary transition, a process during which the numbers of differentiated acinar and β cells increase significantly.4,22 More recent studies have shown that early pancreatic mesenchyme generally induces growth and proliferation of epithelial cells, while at later stages, mesenchymal signals promote exocrine and prohibit endocrine cell differentiation.23,24
INDUCTION OF THE PANCREATIC GENE EXPRESSION PROGRAM
The morphologic changes that occur as pancreas differentiates from gut endoderm depend on sequential changes in gene expression. Extracellular signals provided by tissue interactions between the developing pancreatic endoderm and adjacent tissues ultimately affect cell phenotype by altering gene expression within the nucleus. Recent studies have outlined the underlying molecular events that control these changes in gene expression and have focused in particular on the nuclear proteins, the transcription factors that control gene transcription.
Transcription factors expressed broadly in the endoderm provide competence to respond to the endoderm patterning signals and include several transcription factors originally described in the adult liver, such as hepatic nuclear factors 1b (a POU-homeodomain factor),25 3a and b (forkhead factors now known as Foxa1 and Foxa2),26 4a (an orphan nuclear receptor),27,28 and 6 (a cut-homeodomain factor now known as Onecut1),29,30 and the zinc-finger transcription factors Gata4 and Gata6.31,32 These genes function in a transcriptional hierarchy that not only controls endoderm patterning but also persists in endoderm-derived organs and plays a role in the mature pancreas.33–38 Several of these endoderm factors have been directly implicated in the control of mature pancreatic gene expression (see Fig. 31-5 and Table 31-1).
The earliest genes selectively expressed in prepancreatic endoderm are two transcription factors, the parahox homeodomain factor Pdx139–41 and the basic-helix-loop-helix (bHLH) transcription factor Ptf1a.42,43 Pdx1 expression first appears in prepancreatic endoderm more than a day before the initial formation of the dorsal pancreatic bud, and is preceded immediately by the appearance of another parahox factor, Mnx1 (also known as HB9),44,45 which is expressed more broadly than Pdx1 in anterior endoderm. Expression of both Mnx1 and Pdx1 persists in the initial pancreatic buds, although Mnx1 expression is extinguished quickly, and Pdx1 expression lasts a few more days. Expression of both factors is reactivated in mature β cells.
Dorsal, but not ventral, expression of Pdx1 depends on Mnx1.44,45 Because Mnx1 is also expressed in the notochord during the same period, it is formally possible that Mnx1 could control dorsal Pdx1 expression via signals from the notochord. Pdx1 expression in the dorsal prepancreatic endoderm does not require signals from the notochord, however, suggesting that Mnx1 functions cell intrinsically in inducing Pdx1. Extrinsic signals that induce Pdx1 expression in the dorsal prepancreatic endoderm have not been identified.6 On the other hand, as described in the preceding section, signals from the LPM, including activin, BMPs, and retinoic acid, can induce Pdx1 expression in the ventral prepancreatic endoderm.7
Studies of the Pdx1 promoter have identified several additional endodermal transcription factors as potential intrinsic regulators of Pdx1 expression.46–53 These include members of the Hnf1 and Foxa families of transcription factors, Onecut1, the paired homeodomain factor Pax6, and Pdx1 itself46,47,52,54,55 (Mnx1 also may act through these same Pdx1 binding sites, given its similarity to Pdx1 in the DNA-binding homeodomain). Hnf1a and Pax6 are not expressed early enough or broadly enough to initiate the early expression of Pdx1 in the embryonic gut and pancreatic buds, but the expression patterns of Onecut1, Hnf1β, Foxa1, and Foxa2 suggest that they could play this role (Fig. 31-2). Mice lacking Onecut1 have reduced expression of Pdx1,55 and embryoid bodies lacking Foxa2 fail to activate the pdx1 gene,52 suggesting that these two factors may be bona fide activators of Pdx1 expression in the prepancreatic endoderm and pancreatic buds and therefore lie directly upstream of Pdx1 in the hierarchy of factors involved in initiating pancreas development (see Fig. 31-5).
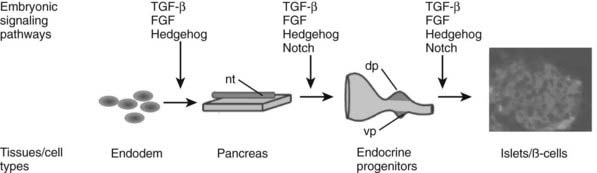
FIGURE 31-2. Embryonic signaling pathways regulate different steps of pancreas and endocrine islet formation. dp, Dorsal pancreas; nt, notochord; vp, ventral pancreas.
Pdx1 is required for outgrowth of the pancreatic buds. In mice with targeted disruptions of the pdx1 gene, the pancreatic buds fail to branch and expand after initial formation, yielding an animal that lacks a pancreas at birth.40,41,56 Confirming its role upstream of Pdx1 in the endoderm, lack of Onecut1 results in reduced pancreatic size,55 and removal of Mnx1 specifically arrests the growth of the dorsal pancreatic bud.44,45 A homozygous null mutation in the IPF1 gene, which encodes human Pdx1, has been identified in a human patient with pancreatic agenesis,57 and mutations in the zebrafish gene encoding Pdx1 also affect pancreas development,58 reflecting an evolutionary conservation of Pdx1 function.
Expression of Ptf1a follows shortly after Pdx1 and is restricted within the endoderm to the dorsal and ventral pancreatic buds.59,60 The aorta induces expression of Ptf1a in the dorsal pancreatic bud, but vascular-derived signals do not appear to play as critical a role in its expression in the ventral buds.20 Although Ptf1a was initially described as an exocrine-specific transcription factor,43,60 in fact it plays an essential role in determining pancreatic cell fate. In mouse embryos lacking Ptf1a, cells normally fated to contribute to both exocrine and endocrine lineages in the pancreas instead revert to duodenal epithelium.42
Cell Type Differentiation
SIGNALING PATHWAYS
Several embryonic signaling pathways regulate distinct aspects of pancreas organogenesis and endocrine cell differentiation. As discussed earlier, TGF-β, FGF, and Hedgehog signaling pathways coordinate initiation of pancreas formation. After bud formation, these pathways play a role in the differentiation of distinct cell types that form the mature pancreas.
FGF7 and FGF10 are expressed transiently in pancreatic mesenchyme and have been shown to promote proliferation of pancreatic epithelial cells.61,62 Transgenic mice that carry a homozygous deletion in the FGF10 gene display severe defects in pancreas morphogenesis that result from loss of Pdx1-positive, pancreatic epithelial progenitor cells.62 FGF signaling also has been shown to regulate proliferation of endocrine and exocrine cells,63,64 and expression of a dominant active form of the FGF receptor 2 (FGFR2) inhibits endocrine and exocrine cells.63–65
TGF-β/activin signaling has been shown to affect differentially exocrine and endocrine cell differentiation. Treatment of isolated pancreatic buds with soluble TGF-β1 stimulates differentiation of endocrine cells, particularly β and PP cells.66 Signaling via activins, a subgroup of TGF-β signaling members, also has been shown to affect preferentially endocrine cell differentiation. Treatment of pancreatic buds with follistatin, an antagonist that physically binds to activins, inhibits endocrine cell differentiation while promoting exocrine cell formation.67 Transgenic mice ectopically expressing dominant-negative activin type II receptor (dnActRII) develop islet hypoplasia, a phenotype also observed in mice carrying targeted mutations in both ActRIIA and ActRIIB.68–70 More recent work has focused on the downstream signaling cascade of TGF-β/activin signaling. Ligand binding to their respective receptors on receiving cells results in the phosphorylation of receptor-associated Sma- and Mad-related proteins 1, 2, 3, 5, and 8 (R-Smads). Another Smad, Smad4, acts as a universal partner for the R-Smads, and the R-Smad/Smad4 complex interacts with distinct DNA-binding factors to regulate target gene transcription. It is surprising that loss of Smad4 has no appreciable effects on pancreas formation,71,72 suggesting Smad4-independent TGF-β/activin functions. Additional support for the TGF-β/activin requirement during pancreas development comes from recent studies in which Smad7, a negative factor that interferes with Smad-receptor and Smad-Smad interactions, is ectopically expressed in pancreatic tissue. Embryonic expression in pancreatic epithelium results in pancreas and β cell hypoplasia.73 Of note, ectopic expression specifically in adult pancreatic Pdx1+ cells resulted in hypoinsulinemia followed by diabetes.
The role of Hedgehog signaling during endocrine cell differentiation is less clear. At early stages, elevation of Hedgehog signaling in the pancreas anlage results in transformation of the pancreatic mesenchyme into duodenal mesoderm.17,74 Increased Hedgehog signaling impairs pancreatic organogenesis, at least in part, because signals normally provided by the pancreatic mesenchyme are missing. Also, as has been shown for other organs, ectopic activation of Hedgehog signaling reduces FGF10 expression known to promote pancreatic epithelial and endocrine cell expansion.75,76 By contrast, a low level of Hedgehog signaling is detected throughout pancreas organogenesis and in mature islets.77 In addition, cell culture experiments have revealed that Hedgehog signaling activates Pdx1 and insulin promoters in cultured insulinoma cells,78,79 suggesting a different and potentially important role for the pathway in maintaining mature β cell function. As they result in early embryonic lethality before pancreas formation is initiated, conventional knockout mice lacking all Hedgehog signaling have not proved useful in determining whether pancreatic Hedgehog signaling is essential for some aspects of pancreas organogenesis.80 These questions await tissue-specific inactivation of the pathway in the pancreas.
Hedgehog signaling shares many features with another embryonic pathway, the canonical Wnt cascade. Several Wnt ligands and their respective frizzled receptors have been identified in embryonic and adult pancreas.81,82 Functional studies in which dominant-negative receptors were expressed ectopically, or β-catenin, an essential component of the canonical Wnt signaling cascade, was eliminated, have pointed to an important role of this pathway during embryonic pancreas formation; however, the exact function of each specific pancreas cell type is still under debate. Although several studies found a requirement for canonical Wnt signaling during exocrine development,83–85 others have noted a role for β-catenin in endocrine function.86 Ectopic activation of β-catenin signaling results in different phenotypes, depending on the time of expression. Activation at the onset of pancreas formation blocks progenitor cell expansion, and later expression results in a profound increase in acinar cell numbers.87 A recent study analyzing the requirement for the mouse homolog of pygopus 2 (mPygo-2), a necessary nuclear component of the Wnt cascade, revealed a novel role for Wnt signaling in early pancreatic mesenchyme.88 According to this study, mesenchymal mPygo-2 is essential in regulating epithelial progenitor proliferation, including endocrine progenitors. Thus, Wnt signaling regulates pancreas formation at various stages in both mesenchyme and epithelium. Furthermore, studies in adult animals indicate a role for canonical Wnt signaling in adult β cell proliferation.89
In addition to the TGF-β, FGF, Hedgehog, and Wnt signaling pathways, Notch signaling contributes to cell fate choices and differentiation in the pancreas, as it does in many other organs during embryogenesis. Notch signaling commonly regulates cell fate decision via a process known as lateral inhibition, in which a given cell within a homogenous field of cells becomes less susceptible to ligand-activated Notch signaling. As a consequence, this cell initiates a specific differentiation program while continuing to provide Notch ligands to adjacent cells, thereby blocking the differentiation of its neighbors (Fig. 31-3).90 A similar mechanism regulates the differentiation of endocrine cells during pancreas development. Reduction of Notch pathway activity in transgenic mice lacking essential Notch signaling components results in upregulation of expression of Neurogenin3 (Neurog3), a bHLH transcription factor required for endocrine formation91 (see following section), and precocious endocrine differentiation.92,93
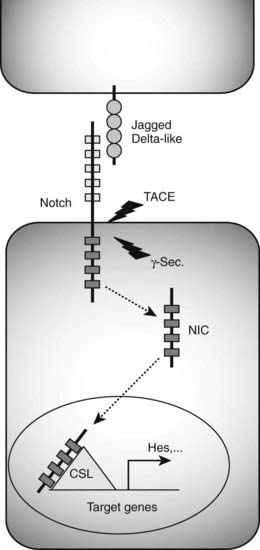
FIGURE 31-3. Notch signaling pathway. Binding of Jagged/Delta-like ligands to Notch receptors activates the Notch signaling cascade. Two proteolytic events mediated by the tumor necrosis factor-α–converting enzyme (TACE) and γ-secretase (γ-Sec.) enzymes result in the generation of an intracellular Notch fragment (NIC) that translocates from the membrane to the nucleus. Interaction with transcription factors from the CSL (CBF1/suppressor of hairless/Lag-1) family activates transcription of Notch target genes, including Hes genes known to block the transcription of Neurog3.
Recent studies have revealed roles for Notch signaling in other steps of pancreatic formation, both before and after its well-recognized role in regulating endocrine specification. Even before pancreas is specified, Notch signaling in the foregut endoderm helps to define the area of the pancreas domain.94 Ectopic expression of a constitutively active, truncated form of the Notch1 receptor at the onset of pancreas formation blocks both exocrine and endocrine cell differentiation, suggesting that Notch signaling normally may allow the expansion of undifferentiated precursor cells.95–97 After endocrine specification, constitutive activation of Notch signaling also blocks endocrine cell differentiation in endocrine precursor cells and can change the differentiation potential of some α/β cells away from the endocrine phenotype toward the ductal fate.98 By contrast, Notch activation in fully matured endocrine cells is not sufficient to change their differentiation status.
Although the exact mechanism of Notch regulation during pancreas formation remains unresolved, recent evidence suggests that FGF signaling moderates Notch activity during pancreas formation. Ectopic expression in the pancreas epithelium of FGF10, an FGF ligand normally found in pancreatic mesenchyme, activates expression of Notch ligands and receptors and the Notch target gene Hes1.99,100 Given the recent evidence of interactions between embryonic signaling pathways during the formation of other organs, it will be critical to determine how these exchanges regulate pancreas development and pancreas cell differentiation.
TRANSCRIPTION FACTORS
The target of notch inhibition in the embryonic pancreas, bHLH factor Neurog3, functions as a proendocrine factor: It is sufficient by itself to drive precursor cells to an endocrine fate.92,101 When expressed broadly in the developing pancreatic bud, Neurog3 can force all cells in the developing pancreas to differentiate prematurely into islet cells. It is interesting to note that Neurog3 is expressed transiently, predominantly during development in the pancreas in scattered ductal cells and occasional periductal cells, but it is never found in mature, hormone-producing cells. Together, these data support a model in which Neurog3 acts upstream of other islet differentiation factors, initiating the differentiation of endocrine cells, but switching off before the time of final differentiation. This conclusion is supported by the observation that mice homozygous for a targeted disruption of the Neurog3 gene fail to form any endocrine cells in the pancreas and do not express the other islet differentiation factors,91 as well as by lineage tracing experiments.102
Although Notch signaling restricts Neurog3 to scattered cells within the pancreatic epithelium, positive signals must initiate Neurog3 expression in the absence of Notch signaling. Studies of the Neurog3 promoter implicate several transcription factors in this role, including Hnf1, Foxa, Onecut1, and Sox9,103–106 which function together as an interacting network105 (Fig. 31-4). Roles for Onecut1 and Sox9 have been confirmed in vivo.104,106
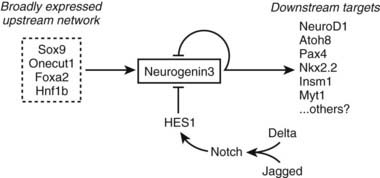
FIGURE 31-4. Control of Neurog3 expression. A model is shown for the positive and negative regulation of Neurog3 expression in pancreatic endocrine progenitor cells.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


