Kallmann Syndrome
The prototype of GnRH deficiency exists in the form of Kallmann syndrome, which refers to the association between hypogonadotropic hypogonadism and anosmia, first described in 1944.49 Sporadic cases and multiple heritable forms have been described, including X-linked, autosomal recessive, and autosomal dominant. The X-linked form of the disease is caused by mutations in the KAL gene on Xp22.3.50 This gene encodes for the extracellular matrix glycoprotein anosmin-1, which is a cell-adhesion molecule essential for normal neuronal migration during embryogenesis.51,52 Highly conserved across species, anosmin-1 appears to have both direct and permissive roles in olfactory-bulb development, GnRH-neuronal migration, and axonal growth.53,54 Numerous mutations within the KAL gene have been described in patients with Kallmann syndrome.55–57 Interestingly, significant clinical heterogeneity in the degree of hyposmia/anosmia and hypogonadism exists among patients bearing identical mutations, implying the presence of important modifier genes and/or environmental factors in the expression of the syndrome.58,59 The specific genes underlying the other forms of Kallmann syndrome are unknown, although loss-of-function mutations in FGFR1 and mutations in prokineticin-2 signaling and in nasal embryonic LH-releasing hormone (LHRH) factor (NELF)60,61 have been reported in association with the autosomal-dominant form.62 A wide range of associated congenital anomalies have been reported in patients with Kallmann syndrome, including cleft palate, hearing loss, color blindness, abnormal eye movements, unilateral renal agenesis, and synkinesia, although the relationship between these features and specific abnormalities of the KAL gene are unclear.63
GnRH Receptor Mutations
The search for GnRH gene mutations has been negative thus far,64 but mutations in the GnRH receptor have increasingly been identified as a cause of familial isolated hypothalamic-pituitary hypogonadism.65 This disorder is transmitted in an autosomal-recessive fashion, affecting both males and females, and olfaction is normal. Several different loss-of-function mutations in the GnRH receptor gene have been described, resulting in a phenotypic spectrum of hypogonadotropic hypogonadism.66–68 Both homozygous and compound heterozygous cases occur, and carriers for the mutations are unaffected.69 As in Kallmann syndrome, pulsatile GnRH administration may restore normal gonadal function and fertility in some patients with partially inactivating GnRH receptor mutations.70
GPR54 Gene Mutations
A novel etiology for autosomal-recessive isolated hypothalamic-pituitary hypogonadism has been discovered in the form of mutations in the GPR54 gene, which encodes an orphan G protein–coupled receptor that is a member of the rhodopsin family of receptors.71 Mutations of the GPR54 gene are less common causes of hypothalamic-pituitary hypogonadism compared to GnRH receptor mutations.72 Several consanguineous families have been identified, as well as one unrelated individual found to be a compound heterozygote for GPR54 mutations.73 The putative ligand of GPR54 is derived from kisspeptin-1, a tumor-suppressor gene.74 Although the frequency of GPR54 mutations appears to be low among kindreds with autosomal-recessive hypothalamic-pituitary hypogonadism, studies from a transgenic mouse model suggest that GPR54 may be a key regulator of hypothalamic GnRH release and pubertal onset.75 There is increasing evidence that loss-of-function GPR54 mutations in humans slow down pubertal maturation of the gonadotropic axis rather than impeding onset of puberty.76 Fertility can be achieved with exogenous GnRH in men and women with GPR54 mutations.77
Mutations in Pituitary Transcription Factors and Orphan Nuclear Receptors
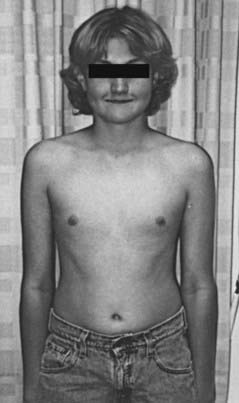
Normal embryologic hypothalamic-pituitary development relies on a tightly regulated, complex cascade of tissue-, cell-, and stage-specific transcription factors. Mutations within the genes encoding for these transcription factors, therefore, may result in a spectrum of combined anterior-pituitary hormone deficiencies and abnormal function of the hypothalamic-pituitary axis. While the etiology of many forms of hypopituitarism remains unknown (Fig. 122-1), molecular genetic abnormalities in several pituitary transcription factors have now been elucidated.
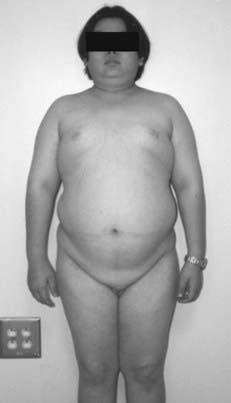
FIGURE 122-1. Delayed puberty in a 19-year-old boy with hypopituitarism of unknown etiology. Bone age was 14 years, and height was 122 cm.
(Reproduced with permission from Becker KL [ed]: Principles and Practice of Endocrinology and Metabolism, 3rd ed. Philadelphia: Lippincott Williams & Wilkins, 2001.)
Pit-1
The first identified transcription factor, pituitary-specific transcription factor (Pit-1), is essential for normal activation of the genes encoding for GH, prolactin, and thyrotropin-stimulating hormone (TSH), as well as for normal differentiation and proliferation of the pituitary cell types responsible for the expression of these hormones.78,79 Following its discovery in 1988, numerous additional transcription factors have been identified, some of which are also involved in normal function of hypothalamic GnRH and pituitary gonadotrophs.80,81 Orphan nuclear receptors that regulate development within multiple levels of the HPG axis are also associated with hypothalamic-pituitary hypogonadism. Human genetic mutations that have been identified in these factors are discussed in the following sections.
Prop-1
Prop-1 (Prophet of Pit-1) is a paired-like homeodomain transcription factor that is required for normal expression of Pit-1.82 PROP1 mutations in humans are associated with GH, prolactin, and TSH deficiency in addition to LH and FSH deficiencies.83 Pubertal progression has been quite variable in reported cases, with several individuals completing pubertal maturation prior to the development of permanent hypogonadism by their early 20s.84,85
LHX3
Another etiology of combined pituitary hormone deficiency has been traced to mutations within LHX3, a pituitary transcription factor gene located on chromosome 9.86 Although a limited number of cases have been identified, all affected individuals have had gonadotropin deficiency and unresponsiveness to exogenous GnRH.87
LHX4
Heterozygous LHX4 mutations are a rare cause of combined pituitary hormone deficiency and are associated with variable degrees of LH and FSH deficiencies.88
HESX1
Several different mutations within the transcription factor HESX1 gene have been implicated in septo-optic dysplasia associated with variable pituitary function. While some patients exhibit apparently normal pituitary hormone secretion, a continuum ranging from isolated GH deficiency to panhypopituitarism has been described.89 Studies from the mouse model have indicated that HESX1 has an important role in embryologic development of the optic nerves and the hypothalamic-pituitary axis.
SOX2
Several heterozygous mutations have been described within the transcription factor SOX2 gene. In addition to anophthalmia or severe microphthalmia, affected individuals have gonadotropin deficiency. Other associated findings include developmental delay, defect of the corpus callosum, esophageal atresia, and sensorineural hearing loss.90,91
DAX1
Hypogonadotropic hypogonadism is a feature of X-linked adrenal hypoplasia congenita. The gene responsible for this condition, DAX1, encodes for an orphan nuclear receptor that is expressed in adrenal, pituitary, hypothalamic, and gonadal tissue.92 More than 100 different mutations within DAX1 have been identified.93 Males with the disorder typically present with adrenal crisis during infancy, whereas the hypothalamic-pituitary hypogonadism emerges in adolescence.94 However, there appears to be significant clinical variability among individuals with adrenal hypoplasia congenita. Congenital gonadotropin deficiency has been suggested by cryptorchidism in some newborns with DAX1 mutations, while a normal mini-puberty of infancy has also been reported in affected boys.95,96 Atypical presentations have included hypothalamic-pituitary hypogonadism in a female with a homozygous DAX1 mutation,97 delayed puberty in heterozygous females,98 and the delayed onset of adrenal insufficiency and hypogonadism in adolescence99 and adulthood in a few affected individuals.100 Studies from the Ahch (Dax1) knockout mouse suggest that DAX1 has a critical role in testis development and spermatogenesis, as well as in the function of the HPG axis.101 This finding, as well as recognition of the combined hypothalamic and pituitary defect in these patients, has been proposed to explain the disappointing results using exogenous GnRH or gonadotropins to induce virilization and fertility.102
SF1
Like DAX1, steroidogenic factor 1 (SF1) encodes for an orphan nuclear receptor that regulates gene transcription in multiple tissues including the gonads, adrenals, hypothalamus, and pituitary.103 The importance of SF1 for normal gonadotrope function was first established by the knockout mouse, which exhibits gonadotropin deficiency and hypoplastic gonads.104 Human SF1 mutations have been identified as a cause of 46,XY sex reversal and adrenal failure.105,106 Some SF1 mutations cause testicular dysgenesis without affecting adrenal function. Ovarian development appears to be normal thus far in a prepubertal girl with a heterozygous SF1 mutation and adrenal insufficiency.107
Mutations in Gonadotropins and Their Receptors
FSH and LH are composed of a common α subunit and a distinct β subunit and exert their biological effects through G protein–coupled receptors (see Chapter 5). No mutations of the α subunits have been described to date, but mutations of the FSH-β subunit, LH-β subunit, and inactivating gonadotropin-receptor have been identified as causes of delayed puberty and abnormal reproductive function in males and females.108 These mutations continue to provide important insight into the relative roles of FSH and LH in gonadal function and human physiology.
FSH-β Subunit Mutations
Mutations in the FSH-β subunit gene have been reported to cause hypogonadism in both females and males.109 The majority of cases have occurred in females with complete absence of pubertal development, although partial breast development in an affected girl has been reported.110,111 The phenotype of males with FSH-β gene mutations ranges from delayed puberty and azoospermia to azoospermia alone.112 As expected, serum LH levels are high in these patients, and FSH is undetectable.
FSH Receptor Mutations
Homozygous inactivating mutations in the FSH receptor were first identified as the cause of autosomal-recessive premature ovarian failure in several Finnish families.113 The females in these kindreds demonstrated uneven degrees of pubertal development, followed by complete ovarian failure in association with elevated serum gonadotropins and hypoplastic ovaries. Subsequently, a number of novel FSH receptor mutations have been described in association with primary or secondary amenorrhea.114,115 In contrast, men with homozygous mutations have normal LH and testosterone levels and low-normal testicular volumes, with elevated FSH levels and unpredictable degrees of impaired spermatogenesis and infertility.116
LH-β Subunit Mutation
One case of delayed puberty due to an LH gene mutation has been described. The 17-year-old male proband was homozygous for a single base substitution in the LH-β subunit gene.117 Serum LH was elevated, while FSH and testosterone levels were low. The finding of LH immunoreactivity with complete loss of LH bioactivity in this patient was consistent with the clinical phenotype. Three maternal uncles, all heterozygous for the same mutation, were infertile. Although female carriers appeared unaffected in this family, anovulatory infertility was reported in a woman with a novel mutation of the LH-β subunit, indicating variable genetic expression in this condition.118
LH Receptor Mutations
Homozygous loss-of-function mutations of the LH receptor, inherited via autosomal-recessive transmission, have been recognized as a rare cause of hypogonadism in males and females. The first such mutation was reported in two 46,XY individuals with female external genitalia but complete absence of breast development and primary amenorrhea.119 Serum levels of LH were markedly elevated, while testosterone levels were low, and FSH levels were normal. Histologic examination of the testes revealed Leydig-cell hypoplasia. Since then, multiple additional mutations of the LH receptor have been identified.120–122 Males with inactivating LH receptor mutations exhibit a clinical spectrum ranging from female external genitalia to hypospadias or micropenis.123 The testes are usually cryptorchid, and müllerian structures are absent. In females, LH receptor mutations result in amenorrhea with normal secondary sexual development and elevations of LH and FSH.124 The finding of ovarian follicular development at all stages suggests that LH may be necessary for ovulation, but that follicular development may occur under the influence of FSH and ovarian paracrine factors alone.125 Conversely, activating mutations of the LH receptor cause a form of male-limited familial precocious puberty and are discussed in Chapter 121.
Metabolism-Related Genetic Defects
Naturally occurring genetic mutations associated with obesity have indicated important interactions between metabolism and human reproduction. Hypothalamic-pituitary hypogonadism has been a feature of rare human mutations within the leptin gene and leptin receptor.126,127 A similar phenotype was observed in a girl with primary amenorrhea and hypothalamic-pituitary hypogonadism in association with compound heterozygous mutations in prohormone convertase 1, an endopeptidase involved in neuropeptide processing.128 The precise role of these genes in GnRH release and/or function is unknown.
Chronic Disease
Any significant chronic disease or inflammation can result in delayed puberty, which is usually seen in association with poor growth and retarded skeletal maturation. Malnutrition also has a profound impact on gonadal function through loss of normal hypothalamic-pituitary pulsatile secretion. This is exemplified by the hypothalamic amenorrhea often observed in young women with anorexia nervosa, in whom chronic caloric restriction leads to aberrations in neuroendocrine function.129 The same phenomenon is observed in individuals engaging in frequent, high-intensity exercise such as runners or ballet dancers, in whom osteopenia is a common correlate.130,131 Hypothalamic-pituitary hypogonadism has also been documented in women of normal weight who practice severe restriction of dietary fat intake.132 Loss of GnRH pulsatility in these settings is likely the result of leptin deficiency, a hypothesis supported by the restoration of ovulatory cycles following human recombinant leptin administration in women with anorexia nervosa.133 These forms of hypogonadism are transient and characterized by a restoration of normal HPG function once the underlying abnormality is resolved. In contrast, permanent hypothalamic-pituitary hypogonadism may result from acquired central nervous system insults such as infection, cranial irradiation, tumor, or trauma. Paradoxically, children with intracranial abnormalities are at increased risk for the development of precocious puberty (see Chapter 121).
Syndromes
Hypothalamic-pituitary hypogonadism is a common feature of a number of different genetic syndromes with otherwise heterogeneous manifestations. For example, individuals with Prader-Willi syndrome often have hypogonadism secondary to hypothalamic-pituitary dysfunction.134 Other syndromes that share this characteristic include Boucher-Neuhauser,135 Bardet-Biedl,136 and Noonan syndromes.137 Associated abnormalities often include cryptorchidism, micropenis, or both. Hypothalamic-pituitary hypogonadism also occurs in patients with pseudohypoparathyroidism type IA, in association with multiple hormone resistance from a loss-of-function mutation in the GNAS1 gene.138
PRIMARY HYPOGONADISM
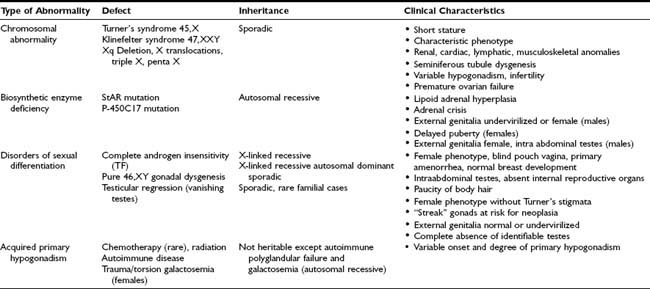
Primary hypogonadism refers to conditions in which there is failure of normal sex-steroid production due to an intrinsic abnormality of the gonads. Using traditional assays, individual gonadotropin levels in prepubertal children with primary hypogonadism may be indistinguishable from normal controls, with the development of elevated gonadotropins by the time pubertal age is reached. Using ultrasensitive methods, supranormal LH and FSH concentrations are detectable in prepubertal agonadal children of all ages compared to controls.139 In contrast, serum inhibin B levels tend to be inversely proportional to gonadotropins in boys with primary gonadal failure.140,141 The causes of primary hypogonadism include congenital and acquired gonadal abnormalities, as discussed in the next section and summarized in Table 122-2.
Congenital Primary Hypogonadism
Chromosomal Abnormalities
It has been recognized for many years that normal ovarian activity depends on the presence and function of specific X chromosomal genetic loci. Therefore, abnormalities involving any significant portion of the X chromosome in females are associated with aberrant ovarian function. Varying degrees of gonadal dysregulation are also observed in males with an abnormal complement of X chromosomes. The most common karyotypic abnormalities leading to primary hypogonadism are discussed in the next sections.
Turner’s Syndrome
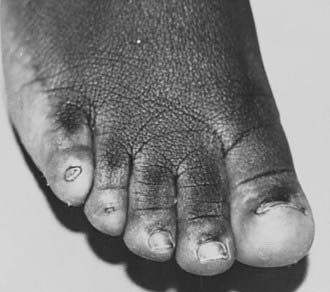
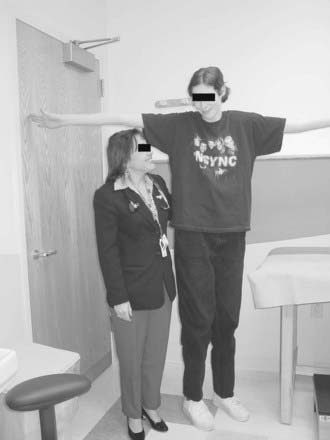
With an incidence of 1 in 2000 to 5000 live-born females, Turner’s syndrome is the most common cause of primary ovarian failure. First described in 1938 by Dr. Henry Turner,142 the syndrome consists of a missing or structurally abnormal X chromosome combined with characteristic phenotypic features. Although a wide range of physical characteristics may manifest, nearly 100% of affected individuals have short stature. Gonadal dysgenesis, as evidenced by “streak ovaries,” is present in approximately 94% of patients and appears to occur as a consequence of accelerated oocyte atresia, with subsequent fibrosis of ovarian stroma.143 The multisystem involvement is demonstrated by the frequent occurrence of cardiac, renal, lymphatic, and musculoskeletal anomalies,144 while common phenotypic features include low-set, posteriorly rotated ears, high-arched palate, low posterior hairline, webbed neck, the appearance of widely spaced nipples, cubitus valgus, and hypoplastic, hyperconvex fingernails and toenails (Figs. 122-2 to 122-4). While many of these somatic characteristics may be the consequence of intrauterine lymphedema, the Turner’s syndrome phenotype in general is thought to result from a haploid dosage of genes on the X chromosome that normally escape X inactivation.145 Girls may come to medical attention in several ways. Lymphedema noted at birth may prompt an early diagnosis. Alternatively, girls may present in midchildhood or adolescence with progressive short stature or delayed puberty. Cytogenetic analysis reveals a heterogeneous group of X chromosomal abnormalities, with approximately 50% of patients exhibiting some form of mosaicism.146 The diagnosis of ovarian failure is based on the finding of elevated gonadotropins. These follow a diphasic pattern, with plasma LH and FSH levels rising during early and late childhood and exhibiting a nadir during middle years, although increased for age.147 FSH levels in young girls with Turner’s syndrome who have mosaic karyotypes are significantly lower than those in girls with monosomy X.148 Although the vast majority of patients will require exogenous sex-steroid replacement, approximately 30% of girls will experience spontaneous pubertal onset, and up to 16% percent will achieve menarche unassisted.149 Rarely, women with Turner’s syndrome are able to conceive, although pregnancies in this population are complicated by a high incidence of fetal loss and congenital anomalies.150,151
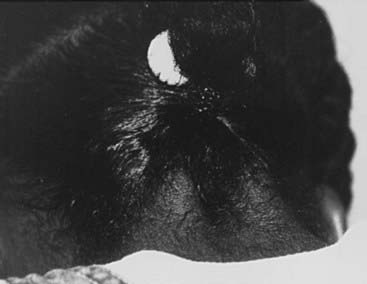
FIGURE 122-2. Girls with Turner’s syndrome often have a low posterior hairline, which typically has a “tripartite” (double arch) configuration.
Xq and Other Chromosomal Abnormalities
Patients with deletions or rearrangements of the long arm of the X chromosome typically have premature ovarian failure without phenotypic features of Turner’s syndrome.152 Molecular genetic analysis has led to the localization within the distal region of Xq of one or more genes essential for ovarian maintenance.153 Many cases of sporadic X chromosome:autosomal translocations are also associated with premature ovarian failure, as are mutations within a forkhead transcription gene (FOXL2) located on chromosome 3.154 Triple and penta-X syndromes represent other examples of X chromosome aneuploidy in which premature ovarian failure is a frequent correlate155 (Fig. 122-5).
Klinefelter Syndrome
First described in 1942,156 Klinefelter syndrome results from X chromosome polysomy in males, with the majority of patients demonstrating a 47,XXY karyotype. It is the most common disorder of the sex chromosomes, with an estimated incidence ranging from 1 in 400157 to 1 in 1000158 live-born males. Typical features include tall stature with eunuchoid body proportions, hypogonadism, a variety of learning and behavioral problems, and borderline-to-normal intelligence.158 The testes, which are usually small and firm and may be cryptorchid, are characterized by seminiferous-tubule dysgenesis159 and a spectrum of Leydig-cell dysfunction. While pubertal onset may occur normally, serum testosterone levels are generally at the low end of the normal range, and inhibin B levels gradually decrease in conjunction with progressive testicular failure.160 Gynecomastia, due to an abnormal estrogen/androgen ratio, and elevated serum gonadotropins are important clues to the presence of Klinefelter syndrome in a boy at pubertal age. However, many patients remain undiagnosed through adolescence161 and may present in adulthood with infertility, azoospermia, or hypogonadism.162 A propensity for the development of certain malignancies has long been recognized in individuals with Klinefelter syndrome. Breast cancer, historically believed to occur at increased frequency,163 has not been observed in other large series,162,164 whereas germ cell tumors have been found to be 50 times more common in this population and have been implicated in rare cases of precocious puberty in boys with Klinefelter syndrome.165
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


