FIGURE 103-1. Adrenal steroidogenesis. Biosynthetic pathways from cholesterol to mineralocorticoids (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione) are shown and the cellular locations of enzyme activities indicated. CMO, Corticosterone methyl oxidase; OH, hydroxylase.
(From White PC, New MI, Dupont B: Medical progress: congenital adrenal hyperplasia, N Engl J Med 316:1519–1524, 1987.
A summary of the clinical, hormonal, and genetic features of these steroidogenic defects appears in Table 103-1.
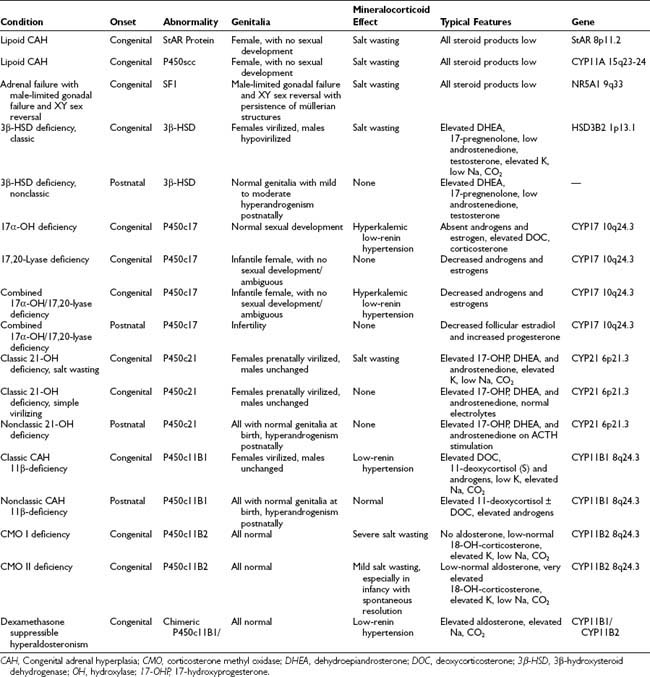
The most common adrenal steroidogenic defects are those related to cortisol production; they are collectively referred to as the CAHs. These defects generally are transmitted as autosomal recessive traits, and the genetic errors that cause them have been described. Loss of the negative feedback inhibition of the hypothalamic-pituitary-adrenal (HPA) axis by cortisol induces oversecretion of adrenocorticotropic hormone (ACTH) by the anterior pituitary, resulting in adrenal hyperplasia. Specific forms of CAH are identified by the abnormal patterns of glucocorticoid, mineralocorticoid, and sex steroid secretion and by accompanying clinical manifestations, including abnormal fetal genital development, disturbances in sodium homeostasis and blood pressure regulation, and the postnatal consequences of sex steroid imbalance affecting somatic growth and fertility. Molecular genetic analysis is used to confirm the diagnosis. Lifelong, carefully monitored treatment with glucocorticoids and salt-retaining steroids can afford many patients with CAH relatively normal lives despite their potentially life-threatening metabolic defects. However, it is not yet possible to always ensure normal hormonal levels throughout the day, ultimate normal stature, and normal fertility.
Distinction is made between classic forms of disease, defined by significantly reduced enzyme activity manifesting clinically at birth, and nonclassic forms, in which the enzyme defect is less severe and symptoms are not present at birth, and when they do appear are generally milder. The classification of CAH subtypes has significant clinical implications for treatment and prenatal diagnosis, but it should be appreciated that patients exist on a clinical continuum rather than within absolute, discrete, easily defined conditions. Approximately 90% to 95% of cases of classic CAH are due to 21-hydroxylase deficiency,1 whereas defects in the enzymes 11β-hydroxylase and 3β-HSD account for almost all the rest. 17α-Hydroxylase deficiency and lipoid CAH are rare causes of CAH.
The 21-hydroxylase and 11β-hydroxylase deficiencies, which occur late in cortisol synthesis, cause shunting of accumulated precursor steroids into pathways of androgen biosynthesis, which do not require these enzymes. Because the external genitalia of the fetus are sensitive to androgens,2 excess androgen secretion by the adrenal masculinizes the female genitalia, causing genital ambiguity in utero in affected females, but no genital alterations in affected males. Postnatal hyperandrogenism affects both genders.
Inefficient/inadequate cortisol production is common to all forms of CAH; imbalances in salt metabolism and fluid volume are part of what differentiates 21-hydroxylase from 11β-hydroxylase deficiency. In 21-hydroxylase deficiency, inadequate aldosterone synthesis leads to salt wasting and hypovolemia, whereas in 11β-hydroxylase deficiency, an excess of mineralocorticoids (e.g., deoxycorticosterone [DOC]) causes expanded fluid volume and hypertension. The nonclassic forms of 21-hydroxylase and 11β-hydroxylase deficiency do not cause severe hypertension or masculinized genitalia in newborn females.
In the 3β-HSD defect, poor conversion of Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, and dehydroepiandrosterone [DHEA]) to Δ4 steroids (progesterone, 17-hydroxyprogesterone [17-OHP], and androstenedione) occurs. The Δ5 steroid precursors are relatively inactive, but the defective cortisol and aldosterone synthesis causes profound salt wasting. Whereas the lack of potent Δ4 androgens produces hypovirilization in the male, enormously high levels of relatively inactive Δ5 androgens, which are converted peripherally to active Δ4 steroids, may cause masculinization of external genitalia in females.
The nonclassic forms of 21-hydroxylase and 11β-hydroxylase deficiencies may be extremely common (and treatable) causes of hyperandrogenism.3–8
In 17α-hydroxylase/17,20-lyase deficiency, blocked production both of 17α-hydroxy (glucocorticoid) and C19/C18 (sex) steroids causes pseudohermaphroditism in males and sexual infantilism in females. Shunting of 17α-hydroxylase precursor steroids into the 17-deoxy pathway produces excess mineralocorticoids (e.g., DOC) and hypertension. Isolated 17α-hydroxylase deficiency prevents the conversion of mineralocorticoids to glucocorticoids, whereas isolated 17,20-lyase deficiency is a variant of 17α-hydroxylase deficiency in which only androgen synthesis is disturbed, and glucocorticoid and mineralocorticoid levels are relatively unaffected.
Lipoid CAH manifests as the inability to produce any of the steroid products from the primary precursor cholesterol. Accordingly, affected individuals are aldosterone, cortisol, and sex steroid deficient. All affected individuals present as phenotypic females, regardless of genetic gender.
History
The observation of hyperplastic adrenal glands in association with internal female gonads and ductal structures in a phenotypic male appeared in the anatomic literature in 1865 with De Crecchio’s9 description of his autopsy of a Neapolitan pseudohermaphrodite. Fibiger,10 Apert,11 and Gallais12 amassed case histories involving precocious puberty, hirsutism, pseudohermaphroditism, and obesity in the early 1900s, and attempted to classify them. The term adrenogenital syndrome was used for many years to describe conditions characterized by elevated adrenal androgens due to virilizing adrenal tumors or to CAH.
The first comprehensive view of CAH was based on the biochemical discoveries of the 1940s and 1950s.13 Among the pioneers who subsequently characterized the variants of CAH in the late 1950s and early 1960s were Bongiovanni,14,15 Eberlein and Bongiovanni,16 Prader and Siebenmann,17 Biglieri and colleagues,18 and New.19 CAH now is more appropriately referred to by the names of the specific enzyme deficiencies that characterize it.
In 1977, the discovery of the association between the well-studied human leukocyte antigens (HLAs) and the 21-hydroxylase trait opened a window to a new form of definition of CAH through molecular genetics.20 Since the gene responsible for classic 21-hydroxylase deficiency (found within the HLA complex) was isolated in 1984,21 knowledge of the specific mutations that cause the different forms of CAH has grown rapidly, much the way that biochemical discoveries of the earlier era led to construction of the scheme for steroidogenesis. Mutations in the genes encoding the steroidogenic enzymes have been confirmed as the basis for all the CAHs. Additionally, steroidogenic defects may be due to mutations in transcription factors or other cofactors (e.g., StAR protein).22
Evidence is accumulating that the correlation between the clinical expression of endocrine disease and mutations of the primary structural gene is not perfect. Thus, the role of the clinician in ascertaining physiologic facts remains central to the prospect of future growth in our understanding of the pathogenesis of CAH and the basis for its treatment.
Disorders of 21-Hydroxylase
Three expressions of 21-hydroxylase deficiency have been clinically identified: classic simple virilizing with manifestations of excess androgen secretion; classic salt wasting, which consists of aldosterone deficiency in addition to excess androgen secretion due to the 21-hydroxylase defect in the parallel mineralocorticoid pathway in the zona glomerulosa; and nonclassic, a less severe hyperandrogenic, variably expressed, and allelically distinct form of the disease, in which affected males and females have unambiguous genitalia at birth.
The result of 21-hydroxylase deficiency is that 17-OHP is not converted to 11-deoxycortisol (compound S) in the pathway of cortisol synthesis, resulting in (1) a deficiency of the essential glucocorticoid cortisol (compound F) and (2) overproduction and accumulation of cortisol precursors proximal to the 21-hydroxylation step (17-OHP, progesterone, 17-hydroxypregnenolone, and pregnenolone) as a result of the loss of normal feedback regulation on the hypothalamus and pituitary. The 17-hydroxylated precursors are shunted into the androgen synthesis pathway. Mutations in the 21-hydroxylase enzyme male may also impair aldosterone synthesis, resulting in the salt-wasting form of CAH.
EPIDEMIOLOGY AND POPULATION GENETICS
Results of newborn screening in a number of localities around the world23–27 yield a worldwide incidence of classic 21-hydroxylase deficiency of approximately 1 in 14,500 live births, ranging from 1 in 858628 to 23,000.29 It has been estimated that 75% have the salt-wasting phenotype.30 Applying the Hardy-Weinberg formula for a population at equilibrium gives a computed heterozygote frequency for classic 21-hydroxylase deficiency of 1 in 61 persons. In areas where patient retesting is difficult/inefficient, it may be necessary to lower the treatment threshold to save lives of affected individuals who die before the diagnosis is confirmed and treatment is started.31
Nonclassic 21-hydroxylase deficiency, one of the most common autosomal recessive diseases, is more frequent than cystic fibrosis. The frequency is ethnic specific, as first determined by Speiser and colleagues.32 Incidences are 1 in 27 Ashkenazi Jews, 1 in 40 Hispanics, 1 in 50 Yugoslavs, 1 in 300 Italians, and 1 in 100 in a heterogeneous New York population.32–34 In an attempt to determine an earliest date for the appearance within the Ashkenazi Jewish population of a founder mutation, DNA analysis was undertaken for representative individuals from the Roman Jewish ghetto, a community already established by the time of the second Diaspora (70 ce). No evidence was seen in Roman Jews of the B14-related nonclassic 21-hydroxylase deficiency mutation, thus suggesting a date after 70 ce for the appearance of this mutation among Ashkenazi Jews.35 Further genetic characterization of this population in terms of its affinity to the general European population and to other Jewish groups continues.36 The nonclassic 21-hydroxylase deficiency mutation can be dated to between 70 ce and the second millennium, based on the significantly higher frequency observed in Ashkenazi Jews compared with Sephardic Jews.
CLASSIC 21-HYDROXYLASE DEFICIENCY
Effects of Hyperandrogenism
External Genitalia
Adrenocortical cell differentiation occurs early in embryogenesis, and although the biochemical schedule of steroid synthesis has not been completely elucidated, it is clear that genital development in the fetus takes place in the setting of active fetal adrenal steroid synthesis. Because differentiation of the external genitalia is sensitive to androgen, excess adrenal androgen produces genital ambiguity in females affected by classic 21-hydroxylase deficiency. In utero masculinization consists of mild to pronounced clitoral enlargement, varying degrees of labioscrotal fusion, and a urogenital sinus with the type and degree of virilization proportional to the onset of hyperandrogenism (Figs. 103-2 and 103-3). The 21-hydroxylase form of CAH is the most common cause of genital ambiguity in females. Every phenotypic male with hypospadias and bilateral cryptorchidism should be considered a female with CAH and should be evaluated immediately for classic 21-hydroxylase deficiency.
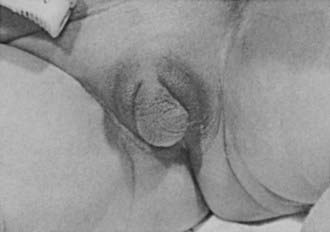
FIGURE 103-2. Ambiguous genitalia in a newborn female with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Note the enlarged clitoris, the single orifice on the perineum, and scrotalization of the labia majora.
(Modified from New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K [eds]: Advances in Human Genetics, vol 4. New York, Plenum, 1973, pp 251–326.)
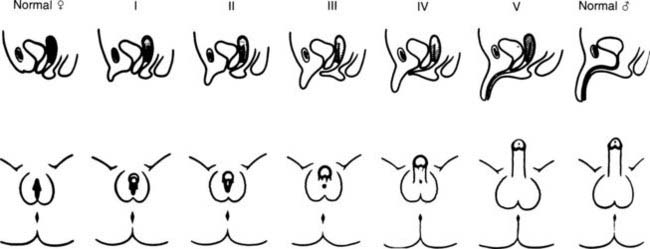
FIGURE 103-3. Prader characterization of the range of genital malformations found in females with classic 21-hydroxylase deficiency. In type I, the only abnormality is enlargement of the clitoris; in type II, partial labioscrotal fusion occurs; in type III, a funnel-shaped urogenital sinus is seen at the posterior end of a small vulva; in type IV, a very small urogenital sinus is found at the base of an enlarged phallus; and in type V, a penile urethra is evident.
(From Prader A: Die Haufigkeit der Kongenitalen Adrenogenitalen Syndromes, Helv Paediatr Acta 13:5, 1958.)
Although differentiation of male genitalia in utero is not affected, the genitalia of infants of both genders undergo androgen-stimulated growth postnatally. High testosterone levels of adrenal origin cause gonadotropin suppression, which results in the characteristic finding in older boys of a large penis and small testes. Hyperpigmentation of the genitalia of both males and females can also result from high ACTH secretion.
Internal Genitalia
Gonadal differentiation and internal genital morphogenesis are not affected by the enzyme abnormalities of classic steroid 21-hydroxylase deficiency. Because there is no anomalous secretion of antimüllerian hormone (AMH), which is synthesized by the Sertoli cells of the fetal testis, müllerian duct development in the female proceeds normally into the uterus and fallopian tubes.37 Thus, normal child-bearing capacity exists in females. Wolffian duct stabilization and differentiation normally take place in the context of local testosterone levels in the male, and this process appears to be unaffected by elevated systemic prenatal adrenal androgens.
Growth
Postnatal somatic growth in both genders is markedly affected by the chronic hyperandrogenism of untreated 21-hydroxylase deficiency. High levels of androgens cause accelerated growth in early childhood, producing an unusually tall and often quite muscular child, an “infant Hercules” (Fig. 103-4). This early growth spurt, however, is followed by premature epiphyseal maturation and closure, resulting in a final height that is short relative to that expected on the basis of midparental target height. Exposure to glucocorticoids used in replacement therapy at doses that may exceed the physiologic requirement has been postulated to be another important factor in the poor growth of these patients.38
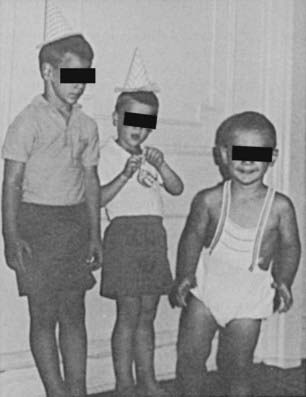
FIGURE 103-4. Untreated congenital adrenal hyperplasia in two brothers: a 4-year-old on the left and a 2-year-old on the right. In the center is their normal 6-year-old brother.
(Modified from New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K [eds]: Advances in Human Genetics, vol 4. New York, Plenum, 1973, pp 251–326.)
We have previously shown that final height is one of the features of CAH least amenable to glucocorticoid replacement therapy.39 Analysis of 47 patients with classic CAH separated into two groups defined by the degree of hormonal control failed to show a significant difference in height outcome with the use of standard treatment.40 A pilot study combining recombinant human growth hormone with a gonadotropin-releasing hormone superagonist to improve final height in those patients with CAH with advanced skeletal maturation and poor predicted final adult height demonstrated that this significantly improved final adult height in children with CAH.41
Hair and Skin Gland Abnormalities
Facial, axillary, and pubic hair often appears very early; pubarche sometimes occurs in infancy. Adult body odor in children, temporal alopecia, and severe acne are other typical features that are reversible with treatment.
Bone Health
Bone mineral density (BMD) is affected by the competing actions of androgen excess (from undertreatment) and glucocorticoid excess (from overtreatment), which can occur in patients simultaneously. The relative extent of undertreatment and overtreatment determines the net effect on BMD. Two studies found that BMD was lower in young adults with CAH than in controls42,43; one of these studies showed a trend toward increased fracture rate of wrist and vertebrae in young women with CAH.43 Dosing regimens, beginning in the young adult years, clearly must take bone health into consideration.
Fertility
Excess adrenal sex steroids inhibit the pubertal changes in gonadotropin secretion directed by the hypothalamic-pituitary axis, probably via a negative feedback effect at the hypothalamus or the pituitary. This inhibition is reversible by suppression of adrenal androgen production. In most untreated or poorly treated adolescent girls and in some adolescent boys, spontaneous pubertal development does not occur until proper glucocorticoid treatment is instituted. Menstrual irregularity and secondary or even primary amenorrhea with or without hirsutism can occur in untreated and poorly controlled women. An association of virilizing CAH with polycystic ovary syndrome has also been noted.44,45 Reproductive capacity in females with classic CAH is reduced. Meyer-Bahlburg46 compared the fertility of women with different forms of classic 21-hydroxylase deficiency. In those women with an adequate introitus and heterosexual activity, he found that fertility was approximately 60% with the simple virilizing form (n = 25), whereas in the salt-wasting form (n = 15), fertility was only 7%.
Traditionally, well-treated patients were thought to have the onset of puberty occur at the expected chronologic age; however, a recent analysis suggests that puberty does in fact occur early than expected—and height is compromised—especially in salt-wasting males.47 The gonadotropin response to gonadotropin-releasing hormone (gonadotropin-releasing hormone or luteinizing hormone-releasing hormone) is appropriate for age in well-controlled prepubertal and pubertal female patients unless ovarian disease is present. Poor control of the disease in males with classic CAH has been associated with small testes and azoospermia. However, cases of normal testicular maturation and spermatogenesis and fertility in untreated men have been reported.48 Well-treated males are normally fertile and have normal pubertal development, spermatogenesis, and testicular function. Later in life, males may develop nodular hyperplasia of adrenal rest tissue, causing enlargement of the testes, which may respond to high-dose glucocorticoid treatment. With the use of ultrasonography, several studies have identified testicular “tumors” in many or even most adolescent and adult male patients with CAH (ages 16 to 40 years).49,50 Tumors ranged in size from 0.2 to 4.0 cm, and in many of these patients, the tumors were palpable. The degree of hormonal control did not correlate with the presence of tumor; indeed, many adequately or even overtreated subjects had identifiable tumors. Genotype was predictive of tumor size, with the most severe mutations correlating with the largest tumors.49 Leydig cell function also was commonly impaired in these subjects.50 The adrenal glands of subjects with mutations in the 21-hydroxylase gene, both homozygous and heterozygous, are known to be larger than those of controls. This increased surface area has been associated with an increased risk of adrenal tumor formation, and indeed adrenal “incidentalomas” are more common in these groups.51 However, a retrospective analysis of “true” adrenal tumors failed to demonstrate an increased incidence of mutations, either germline or somatic.52
SALT WASTING
Salt wasting is characterized by hyponatremia, hyperkalemia, metabolic acidosis, inappropriate sodium excretion in the urine, and low serum aldosterone with concomitantly high plasma renin activity (PRA).
Salt wasting results from inadequate secretion of salt-retaining steroids, especially aldosterone. In addition, hormone precursors elevated in 21-hydroxylase deficiency may act as mineralocorticoid antagonists (e.g., 17-hydroxyprogesterone). Newborns are especially prone to the development of a salt-wasting crisis because the sodium-conserving mechanism of their renal tubules is only marginally competent. The adrenal medulla of patients with CAH may be incompletely formed in some patients. Merke and coworkers53 noted that the epinephrine and metanephrine concentrations of subjects manifesting a salt-wasting crisis were approximately 50% of those of controls; pathologic evaluation of adrenal medullary tissue (removed during adrenalectomy) demonstrated depletion of secretory vesicles.
A single enzyme is responsible for 21-hydroxylation in both the zona fasciculata and the zona glomerulosa. The pathogene-tic difference between salt wasting and simple virilizing 21-hydroxylase deficiency may simply be the result of a quantitative difference in enzyme activity caused by conformational changes induced by the specific mutations. In vitro expression studies show that as little as 1% of normal activity of 21-hydroxylase allows adequate aldosterone synthesis to prevent significant salt wasting.54,55 The issue is complicated by the occasional finding of discordance for salt-wasting expression among siblings with identical mutations in their inherited 21-hydroxylase genes, and by the observed improvement in salt wasting in some individuals over time.48,56–58 In one case, a girl born with male genitalia and salt wasting, presumably with severe enzyme deficiency, was no longer a salt waster by age 4. Another important case finding was that of a girl with a homozygous deletion of the 21-hydroxylase gene and a history of multiple salt-wasting crises in infancy, who discontinued her therapy in adolescence and was found at that time to be secreting aldosterone.59
Salt-Wasting Crisis
Infants with classic CAH of both genders are at risk for hypovolemic and hypoglycemic adrenal crises, typically occurring within the first 4 to 6 weeks of life. Undiagnosed males who lack the genital ambiguity that usually calls attention to the condition in a newborn female are at added risk for shock and death, as evidenced by the higher female-to-male ratio of salt-wasting adults. In females, the degree of genital ambiguity in a newborn does not indicate the likelihood of the potential for salt wasting. Thus, surveillance of infants of both genders in the newborn period for incipient adrenal crisis is essential, and close surveillance should continue after discharge from the hospital. Infants should remain in the hospital after birth for approximately 7 days or until the aldosterone/sodium balance status has been established, although it should be stressed that the first salt-wasting crisis may not occur for several months. In older children and adults, acute adrenal insufficiency can be precipitated by physical exertion or emotional stress, undertreatment, or noncompliance with therapy, or can occur as a result of immunization, infection, trauma, or surgery. The salt-wasting crisis of the newborn period associated with CAH often is confused with pyloric stenosis, which may present at the same age and also often manifests as an ill-appearing newborn with vomiting and dehydration. The salt-wasting crisis associated with CAH (manifesting with hyponatremia, hyperkalemia, and metabolic acidosis) should be differentiated immediately from that of pyloric stenosis (hypokalemia, hypochloremia, and metabolic alkalosis) with the use of routine biochemical tests.
NONCLASSIC 21-HYDROXYLASE DEFICIENCY
Nonclassic 21-hydroxylase deficiency is distinguished from the classic form symptomatically by its age at onset and biochemically by its less severe impairment of steroid 21-hydroxylation; because of these two factors, it does not result in ambiguous genitalia in the newborn female. It often initially manifests just before the age of normal puberty, although it may present at any age after birth. Patients may be homozygous for a mild genetic defect or may have compound heterozygotes for one severe mutation and one mild mutation. Nonclassic 21-hydroxylase deficiency was first defined in the course of family studies of patients with classic CAH. Family members who should have been carriers for 21-hydroxylase deficiency were found to have overt hormonal disturbances that were associated with distinct alleles at the 21-hydroxylase locus. These alleles were determined to be in linkage disequilibrium with the HLA locus on chromosome 6. It thus was found through these studies that the gene locus for 21-hydroxylase lies between HLA-B and HLA-DR.60
In addition to linkage of the 21-hydroxylase locus with the neighboring HLA-B and HLA-DR antigen loci, 21-hydroxylase deficiency alleles are found in linkage disequilibrium with other HLA genes or haplotypic combinations that may include specific alleles of the neighboring genes C4A and C4B encoding the fourth component of serum complement.61 The two most prominent such cases are linkage disequilibrium of the extended haplotype HLA-A3,Bw47,DR7 with the classic salt-wasting form, and HLA-B14,DR1 with nonclassic 21-hydroxylase deficiency.62
Clinical Features
The clinical features of nonclassic 21-hydroxylase deficiency range widely, begin at any age, and commonly wax and wane over time. Although genital ambiguity in the newborn period is never a feature of this disorder, the appearance of any of the other signs and symptoms of hyperandrogenism can prompt the patient to seek medical attention, or patients may be identified through family or population screening. Individuals commonly present just before the time of expected pubarche. Underlying biochemical abnormalities are always demonstrable at any age by ACTH stimulation testing, regardless of whether symptoms are evident.63
Nonclassic 21-hydroxylase deficiency can result in the premature development of pubic hair (pubarche), which may occur as early as 6 months of age.64 In one study, nonclassic 21-hydroxylase deficiency was found in 30% of children with premature pubarche,65 whereas a lower prevalence was reported by another group.66 Severe cystic acne refractory to oral antibiotics and retinoic acid has been associated with nonclassic 21-hydroxylase deficiency by some.67,68 In one young woman, male pattern alopecia was the sole presenting symptom. Menarche in females can be normal or delayed, and secondary amenorrhea or oligomenorrhea is frequent. Final height, as in classic 21-hydroxylase deficiency, is less than predicted based on the midparental target height and linear growth percentiles, even though excess androgen secretion may not have been apparent.40,50
A number of women with polycystic ovary disease have been found on ACTH testing to have a primary adrenal defect, for example, nonclassic 21-hydroxylase deficiency, 3β-HSD deficiency, or 11β-hydroxylase deficiency.45,69–72 The reported prevalence of nonclassic 21-hydroxylase deficiency as a cause of these endocrine symptoms in women ranges from 1.2% to 30%. The scope of the range may relate to differences in the ethnic makeup of the groups studied. Women heterozygous for a 21-hydroxylase gene mutation have been shown to have higher free testosterone levels than controls (i.e., hyperandrogenemia), but, it is important to note, not an increased incidence of hyperandrogenism.73
In boys, presenting signs include early beard growth, pubarche, acne, and an accelerated growth spurt. In men, signs of androgen excess are difficult to perceive, and the manifestations of adrenal androgen excess may be limited to short stature or oligospermia and diminished fertility from adrenal sex steroid–induced gonadal suppression.
Fertility in Nonclassic 21-Hydroxylase Deficiency
It has been recognized for 30 years that infertility in women may be reversed by glucocorticoid therapy. In one report, five patients with irregular menses and high 17-keto(oxo)steroids (urinary androgen metabolites) resumed regular menses and demonstrated adequate suppression of 17-ketosteroids and pregnanetriol (a urinary metabolite of 17-OHP) within 2 months after beginning therapy with glucocorticoids alone, suggesting the presence of an adrenal 21-hydroxylating defect.74 In another report of 18 infertile women with acne and/or facial hirsutism and hormonal criteria consistent with 21-hydroxylase deficiency, seven conceived shortly after initiating prednisone treatment; an additional four women conceived within 2 months of the addition of clomiphene to the prednisone.75 Hormonal profiles after initiation of therapy were not reported in this study. As mentioned earlier, oligospermia and subfertility have been reported in men with nonclassic 21-hydroxylase deficiency.76 We recommend screening for nonclassic 21-hydroxylase deficiency during every evaluation for infertility.
Molecular Genetics
The molecular genetics basis of 21-hydroxylase deficiency has been studied extensively. The 21-hydroxylase enzyme is a microsomal cytochrome P-450 enzyme termed P450c21. The structural genes encoding P450c21, CYP21, and a pseudogene, CYP21P, are located on chromosome 6p21.3, adjacent to the genes C4B and C4A, encoding the two isoforms of the fourth component of serum complement in the class III region of the HLA complex.77,78 CYP21 and CYP21P each contains 10 exons. Their nucleotide sequences are 98% identical in exons and approximately 96% identical in introns.79
Many of the mutations known to cause 21-hydroxylase deficiency are apparently the result of either of two types of recombination between CYP21 and CYP21P: (1) chromatid misalignment and unequal crossing over, resulting in large-scale DNA deletions, and (2) gene conversion events that result in the transfer to CYP21 of smaller scale deleterious mutations normally present in the CYP21P pseudogene. Seven of the eight possible exonic differences between CYP21 and CYP21P have been observed and confirmed to be causative of 21-hydroxylase deficiency. At least 25 mutations causing 21-hydroxylase deficiency have been identified; one single-nucleotide mutation altering the splicing of an intron is particularly common and is associated with both simple virilizing and salt-wasting phenotypes (Fig. 103-5). Deletions and gene conversion events may be common findings in 21-OHD, owing to the chromosomal arrangement of the gene and pseudogene (i.e., homologous genes in a tandem array),80,81 and to the presence of multiple chi-like sequences.82
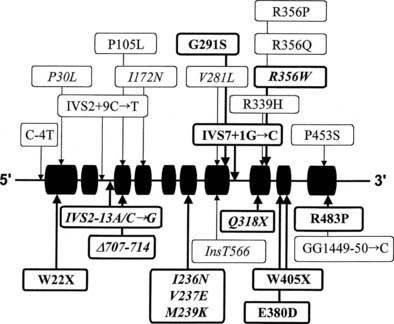
Correlation of Genotype With Phenotype
In general, the functional consequence of each DNA alteration corresponds with the clinical severity of the inherited disease: total deletion of the functional gene, stop codon (nonsense) mutations, frameshifts, and several amino acid substitutions (missense mutations) have been shown to result in salt-wasting classic alleles; one nonconservative amino acid substitution has been associated exclusively with simple virilizing disease, and a single conservative amino acid substitution (V281L) is the mutation associated with the HLA-B14,DR1 haplotype found so frequently in nonclassic disease.83
However, comparison of the clinical characteristics and molecular genetics data in 532 affected individuals at one site reveals that the genotype correctly predicted the phenotype in 89% of cases, a high figure but significantly shy of the 100% that might be expected. Rocha et al.84 suggested that much of the variability in the external virilization of CAH women is due to the genotype (i.e., number of CAG repeats) of the androgen receptor.84 This analysis underlines the need for careful ongoing clinical assessment of all persons affected by 21-hydroxylase deficiency.
Diagnosis
The diagnosis of steroidogenic enzyme defects in CAH is made on the basis of (1) clinical findings, (2) biochemical and hormonal values, and (3) molecular genetics analysis of mutations. Hormonal values will establish the diagnosis of an enzyme defect by demonstrating an increased precursor-to-product ratio. The substrate of the enzyme will be markedly increased, whereas the product will be normal or slightly decreased. Molecular genetic analysis of the DNA will demonstrate the specific mutations.
Biochemical Characterization
In classic CAH, baseline serum cortisol levels are at the lower limits of detection or in the low to normal range. Baseline concentrations of serum 17-OHP and adrenal androgens are elevated, with serum 17-OHP levels often several hundred times normal.85 In nonclassically affected persons, because of pulsatile and diurnal variations in 17-OHP secretion, midmorning and afternoon concentrations may be normal.86 Of all single measurements, early morning measurement of serum 17-OHP concentration is the most likely to show an elevation. However, we caution against relying on baseline 17-OHP levels to exclude nonclassic 21-hydroxylase deficiency; mild defects may be only minimally elevated, especially in the afternoon, leading to many false-negative diagnoses.
The standard diagnostic procedure for diagnosing all forms of CAH is the 60 minute (synthetic) ACTH stimulation test. At 08:00 hours, when cortisol secretion is at its normal diurnal peak, blood is drawn before and then 60 minutes after intravenous injection of a 0.25 mg bolus of ACTH. A nomogram (Fig. 103-6) that relates baseline to ACTH-stimulated serum concentrations of 17-OHP has been constructed that can be used to identify individuals with classic and nonclassic forms, as well as heterozygote carriers of the 21-hydroxylase deficiency form of CAH.
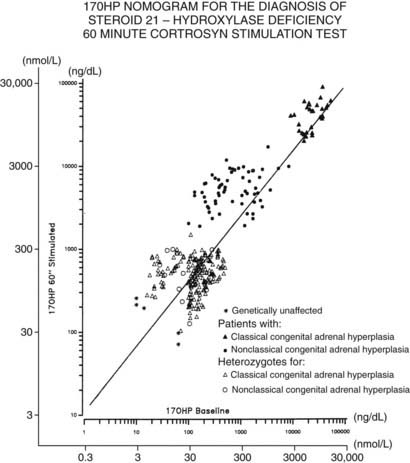
FIGURE 103-6. Nomogram relating baseline to adrenocorticotropic hormone–stimulated serum concentrations of 17-hydroxyprogesterone. Scales are logarithmic. The regression line shown is for all data points. Data points cluster as shown into three nonoverlapping groups: classic (congenital adrenal hyperplasia) and nonclassic forms of 21-hydroxylase deficiency are readily distinguished from each other and from heterozygote/unaffected. Distinguishing unaffected from heterozygote responses is difficult.
Neonatal screening via heel-stick capillary blood has been available since 197787 and is mandated in many states in the United States. The 17-OHP content of the dried blood, which is sampled on filter paper, analogous to the phenylketonuria test standard for all newborns, can be determined by a qualified laboratory. The value of newborn screening has been demonstrated repeatedly by the finding that most (biochemically proven) affected children were not identified on clinical grounds.88–90
The 08:00 hours salivary level of 17-OHP correlates extremely well with serum assays and is highly recommended as a screening test.86 It is especially useful when venipuncture is difficult.
Diagnosis at Birth
The following data should be collected in the evaluation of a newborn with possible CAH:
Further characterization of DNA by molecular genetic analysis is desirable when available.
Disorders of 11β-Hydroxylase
Steroid 11β-hydroxylase activity in the adrenal cortex is required for the synthesis of both glucocorticoids and mineralocorticoids. It has been shown that distinct isozymes of P450c11 participate in cortisol and aldosterone synthesis in humans.91 The structure of the two CYP11B genes (nine exons and eight introns) is also identical to that of the CYP11A gene, which encodes the cholesterol desmolase protein. The genes for the two CYP11B isozymes are 93% identical in predicted amino acid sequence and 90% identical in the intronic region and are encoded by two vicinal genes on chromosome 8q24.3.92–94 It is important to note that the upstream regions of the two genes are quite different, suggesting functionally different control. Disorders of the two enzymes may manifest as hypocortisolism (i.e., CAH), hypoaldosteronism, or hyperaldosteronism.
11β-HYDROXYLASE DEFICIENCY CONGENITAL ADRENAL HYPERPLASIA
Abnormal steroid secretion attributed to defective 11β-hydroxylation was first described by Eberlein and Bongiovanni16 in 1955. It has proved to be the second most common form of CAH (5% to 8% of all cases in the general population).95 In the cortisol pathway, conversion of 11-deoxycortisol to cortisol is reduced. As a result, 11-deoxysteroids (11-deoxycortisol and 11-DOC) accumulate. The hyperandrogenism resulting from shunting of cortisol precursors is similar to that of 21-hydroxylase deficiency, including genital ambiguity in classically affected females.
Approximately two thirds of patients with 11β-hydroxylase deficiency become hypertensive, with or without hypokalemic alkalosis, sometime early in life. Hypertension does not correlate with the presence or degree of hypokalemia or with the extent of virilization.96
In addition to the classic form (i.e., present at birth), milder nonclassic forms of 11β-hydroxylase deficiency have been reported3–8 and may represent allelic variants, analogous to 21-hydroxylase deficiency. No biochemical defect has been demonstrated consistently in the obligate heterozygote parents of patients with the classic form, either in the baseline state or with ACTH stimulation.97 As noted earlier, such a biochemical defect can be demonstrated in 21-hydroxylase heterozygotes. As in 21-hydroxylase deficiency, persons with identical 11β-hydroxylase mutations may differ in the severity of their signs and symptoms of androgen and mineralocorticoid excess, suggesting a role for epigenetic or nongenetic factors in the expression of clinical phenotype.98
Hypertension in 11β-Hydroxylase Deficiency
Hypertension in 11β-hydroxylase deficiency CAH is commonly attributed to DOC-induced sodium retention, which results in volume expansion. However, it has not been consistently proven that DOC causes hypertension.91 In 1970, New and Seaman99 showed that the large amounts of DOC produced in 11β-hydroxylase deficiency are glucocorticoid (dexamethasone) suppressible and therefore originate in the zona fasciculata, rather than in the zona glomerulosa. When DOC is suppressed with dexamethasone treatment, renin levels rise, causing secretion of aldosterone in the zona glomerulosa, in which the 11β-hydroxylase enzyme (P450c11B2) is normal.
Suppression of DOC by glucocorticoid treatment, however, may not lower blood pressure in hypertensive 11β-hydroxylase–deficient patients. Normotensive patients with markedly elevated DOC levels,100,101 hypertensive patients with normal or only mildly elevated DOC,102,103 and atypical 11β-hydroxylase–deficient patients with normal PRA104 present a challenge to the DOC-centered explanation of hypertension. Of course, the lack of response to treatment is a feature of long-standing hypertension of many causes.
Epidemiology
Classic 11β-hydroxylase CAH (due to defects in the CYP11B1 gene) occurs in approximately 1 in 100,000 births in the general white population.105 A large number of cases have been reported in Israel. The incidence there now is estimated to be 1 in 5000 to 1 in 7000 births, with a gene frequency of between 1 in 71 and 1 in 83.106 This unexpected clustering of cases was traced to Jewish families of North African origin, particularly from Morocco and Tunisia. Turkish Jews also have been found to carry the identical 11β-hydroxylase gene mutation with high frequency.96,105 The incidence of the nonclassic form is not known at present but would be predicted to be considerably higher than the classic form, as in 21-hydroxylase deficiency.
Molecular Genetics
One of the 11β-hydroxylase genes, CYP11B1, is expressed at high levels in normal adrenal glands.93 Transcription of this gene is regulated by cyclic adenosine monophosphate (the second messenger for ACTH). Low levels of transcripts of the second gene, CYP11B2, have been detected in normal adrenal with the use of reverse transcriptase polymerase chain reaction. The enzymes encoded by the CYP11B1 and CYP11B2 genes have been studied by expressing the corresponding complementary DNA in cultured cells and after actual purification from aldosterone-secreting tumors.107–109 The high degree of similarity in both gene and protein structures between CYP11B1 and CYP11B2 suggested that the two 11β-hydroxylase genes were members of the same gene family.93,110
The isozyme encoded by CYP11B2, termed P450c11B2, 11β-hydroxylates 11-DOC to corticosterone and 11-deoxycortisol to cortisol. It also 18-hydroxylates corticosterone and further oxidizes 18-hydroxycorticosterone to aldosterone, the latter steps referred to as CMO I and CMO II activity, respectively. The CYP11B2 promotor is sensitive to renin/angiotensin. P450c11B2 is also referred to as CMO I/CMO II and as P450c18 and aldosterone synthase.
In contrast, the product of CYP11B1, termed P450c11B1, has strong 11β-hydroxylase activity but 18-hydroxylates only approximately one-tenth as well as P450c11B2. P450c11B1 does not synthesize detectable amounts of aldosterone from 18-hydroxycorticosterone. The CYP11B1 promotor is responsive to ACTH/cyclic adenosine monophosphate.
These data suggest that P450c11B1 predominantly synthesizes cortisol in the zona fasciculata, whereas P450c11B2 predominantly synthesizes aldosterone in the zona glomerulosa. This hypothesis has been confirmed through the study of individuals with defective cortisol or aldosterone synthesis due to deficiencies in 11β-hydroxylase and CMO II activities, respectively.
Mutations in the zona fasciculata enzyme (P450c11B1) result in defective synthesis of cortisol, producing hypertension from precursor (e.g., DOC) accumulation. Mutations in the zona glomerulosa gene (P450c11B2) result in defective synthesis of aldosterone and consequent salt wasting. An interesting molecular defect of the 11β-hydroxylase genes results in glucocorticoid-responsive hyperaldosteronism and resultant hypertension (see “Dexamethasone [Glucocorticoid]-Suppressible Hyperaldosteronism”).
Almost all affected alleles in the Moroccan-Israeli population carry the same mutation, the substitution of histidine for arginine at codon 448111 (R448H) in the CYP11B1 gene. This mutation is incompatible with normal enzymatic activity112 and appears to represent a founder effect. At least 31 mutations causing 11β-hydroxylase deficiency CAH have been identified and are shown in Fig. 103-7. Mutations are spread across the gene, including at several identified hot spots.113
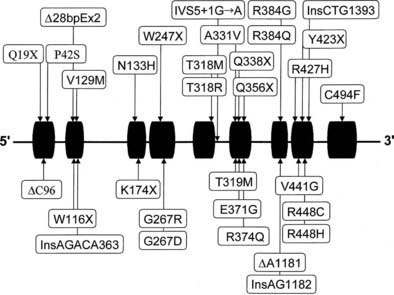
Diagnosis
Elevated serum 11-deoxycortisol (compound S) and DOC, confirmed by marked elevation of their urinary tetrahydrometabolites, is diagnostic. Further confirmation can be found in a complete absence of any 11-oxygenated C19 or C21 steroids in blood or urine.114 Diagnosis is made as for 21-hydroxylase deficiency with the additions of baseline and ACTH-stimulated serum 11-deoxycortisol (compound S) and DOC. Further characterization by molecular genetics analysis should be pursued in families anticipating additional children, with prenatal diagnosis now also available. Diagnosis in a newborn is quite difficult because the characteristic hypertension generally does not appear during the newborn period, and distinction from 21-hydroxylase deficiency based on steroid patterns is also problematic at this age.115 Infantile gynecomastia has been described and may offer a clue to the diagnosis.116 Treatment consists of glucocorticoid replacement. Determination of PRA is the standard test for mineralocorticoid excess. Although a nonelevated DOC is desirable, follow-up usually is accomplished more easily by following the renin (or PRA) level. Conversely, suppressed renin is typically a sign of insufficient control of 11β-hydroxylase deficiency.
CORTICOSTERONE METHYL OXIDASE DEFICIENCY
The conversion of corticosterone to 18-hydroxycorticosterone is referred to as CMO I activity, whereas the subsequent 18-oxidation to aldosterone is known as CMO II activity. In the zona glomerulosa, these steps are catalyzed by distinct domains within a single enzyme, P450c11B2, also referred to as P450c18, aldosterone synthase, and P450aldo. Deficient CMO I activity typically results in (practically) no aldosterone production and phenotypically is more severe than CMO II deficiency. Defects in CMO I activity have been reported but appear to be less common than those of CMO II.117,118 Defects in both CMO I and CMO II activity are inherited as autosomal recessive conditions, and because cortisol synthesis is not affected in either condition, defects of P450aldo are not considered to be forms of CAH.
Infants with CMO II deficiency are subject to potentially fatal electrolyte abnormalities; in childhood, however, recurrent dehydration, a variable degree of hyponatremia and hyperkalemia, and poor growth are characteristic.119,120 Adults may be asymptomatic, their affected status coming to light only in the course of family studies of a symptomatic child. CMO II deficiency has been found at an increased frequency among Jews of Iranian origin.121 Molecular genetic analyses of such families have demonstrated two missense mutations in the CYP11B2 genes of affected individuals: the replacement of arginine by tryptophan at position 181 (R181W) and the replacement of valine by alanine at position 386 (V386A).122
CMO I deficiency is characterized biochemically by essentially unmeasurable aldosterone levels with low or normal 18-hydroxycorticosterone levels. Patients with CMO II deficiency typically have normal (or low) aldosterone levels with elevated 18-hydroxycorticosterone levels (i.e., an elevated serum 18-hydroxycorticosterone-to-aldosterone ratio), along with a low corticosterone-to-18-hydroxycorticosterone ratio. Alternatively, CMO II deficiency can be identified by an elevated ratio of the major metabolites of these steroids (tetrahydro-18-hydroxy-11-dehydrocorticosterone and tetrahydroaldosterone) in urine.120
DEXAMETHASONE (GLUCOCORTICOID)-SUPPRESSIBLE HYPERALDOSTERONISM
Dexamethasone-suppressible hyperaldosteronism (DSH; also known as glucocorticoid-remediable aldosteronism) is a rare form of hypertension123,124 in which aldosterone synthesis appears to be regulated abnormally by ACTH.125 Dexamethasone-suppressible hyperaldosteronism is characterized by (1) rapid and complete suppression of aldosterone secretion on dexamethasone administration, (2) continued increase in aldosterone with long-term administration of ACTH, (3) suppressed PRA, and (4) a dominant mode of inheritance.126 Urinary excretion of 18-hydroxycortisol and 18-oxocortisol is increased.127,128
Individuals with DSH have been shown to have an intergenic recombination, juxtaposing the promoter of CYP11B1 with the coding sequence of CYP11B2.129,130 The fusion or chimeric gene creates an alteration in the regulation of synthesis of aldosterone, making the zona glomerulosa sensitive to ACTH rather than to renin-angiotensin, resulting in glucocorticoid-responsive hyperaldosteronism and hypertension.
Disorders of 3β-Hydroxysteroid Dehydrogenase
The enzyme 3β-HSD is necessary for the synthesis of bioactive Δ4 adrenal and gonadal steroids from the relatively inactive Δ5 precursors. In addition to 3β-hydroxysteroid dehydrogenation, this enzyme performs 3-oxosteroid isomerization, giving rise to the alternate term of Δ4/Δ5 isomerase. 3β-HSD is present not only in the adrenal cortex, gonads, and placenta but also in the liver and in nearly all peripheral tissues. The placental form is distinct from the adrenal/gonadal form. Differences in the levels of 3β-HSD activity in different tissues suggest tissue-specific isoforms or tissue-specific regulation of one or more shared isoforms of this protein.131–134
CLASSIC 3β-HYDROXYSTEROID DEHYDROGENASE DEFICIENCY
3β-HSD deficiency is an autosomal recessive form of CAH that was first described by Bongiovanni135 in 1962. The exact frequency of this rare disorder is unknown. It has been postulated that individuals with defects early in the pathway of steroidogenesis, which severely impair cortisol synthesis (i.e., 3β-HSD and StAR), have poor survival rates.
Owing to reduced 3β-HSD enzyme activity in the gonads, genetic males are incompletely masculinized and exhibit genital ambiguity at birth. In affected females, conversely, very high levels of circulating DHEA converted peripherally to active androgens produce a limited androgen effect. Clitoral enlargement occurs and, rarely, labial fusion. As with the 21-hydroxylase and 11β-hydroxylase enzymes, the severity of the enzyme defect may not be determined on the basis of the appearance of the external genitalia at birth. Deficient steroid production in 3β-HSD deficiency may result in salt wasting, although this clearly is not true in every case.136,137
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


