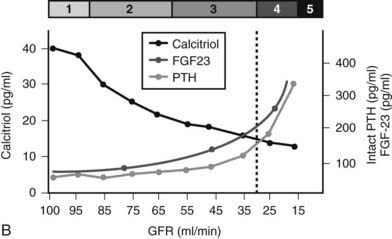
FIGURE 69-1. A, Median levels of calcium, phosphorus, and parathyroid hormone (PTH) per stage of chronic kidney disease (CKD). Median serum levels of calcium and phosphorus stay constant until late in the course of CKD; PTH levels rise before any changes are seen in calcium and phosphorus.
B, In patients with CKD, serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations occur and prior to any rise in serum PTH levels.
(A, Data from Levin A, et al: Kidney Int 71:31–38, 2007.3)
Fibroblast Growth Factor-23
A recently described phosphaturic hormone, FGF-23, was first identified in patients with tumor-induced osteomalacia7 and autosomal dominant hypophosphatemic rickets.8 In these conditions and in patients with X-linked hypophosphatemia, elevated circulating levels of FGF-23 result in renal phosphate wasting and suppression of 1,25(OH)2D3 production.8,9 FGF-23 is made within bone,10,11 and the presence of a cofactor, Klotho, is essential for its action.12 Serum values increase as CKD progresses, becoming markedly elevated in individuals with end-stage kidney disease (Fig. 69-1B).13,14 In patients with CKD, 1,25(OH)2 D3 levels are inversely related to levels of circulating FGF-23, suggesting that increases in FGF-23 early in the course of CKD may play a role in declining active vitamin D levels.14 FGF-23 levels increase in response to vitamin D sterol therapy,15 and levels may be regulated by phosphorous intake.16,17 FGF-23 levels have been implicated in parathyroid gland regulation (vide infra).18,19
Vitamin D
Serum 1,25(OH)2D3 levels decline early in the course of kidney dysfunction, before any changes in serum calcium or phosphorous concentrations and prior to any rise in serum PTH levels.3,20 In late stages of CKD, phosphate retention and increased serum phosphorous levels directly suppress 1α-hydroxylase activity.16 However, although these factors contribute to declining 1α-hydroxylase activity, current evidence demonstrates that rising FGF-23 levels may be of even greater importance.14
Low circulating levels of 1,25(OH)2D3 have consequences for many tissues. Aside from its effect on intestinal calcium absorption, 1,25(OH)2D3 plays a direct role in the suppression of PTH gene transcription (vide infra). Animal studies also indicate that 1,25(OH)2D3 is essential for normal skeletal physiology—particularly in growing animals—and that this effect may not be mediated by the vitamin D receptor (VDR). Mice who lack the VDR [i.e., mice unable to respond to the actions of 1,25(OH)2D3 through its classical receptor] are phenotypically similar to those lacking the 1α-hydroxylase gene itself [i.e., mice unable to generate 1,25(OH)2D3]; both sets of mice are hypocalcemic with markedly elevated serum PTH levels, parathyroid gland hyperplasia, and rickets.21 However, a diet replete in calcium, phosphorus, and lactate is sufficient to normalize serum calcium, phosphorous, and PTH levels and to prevent the development of rickets in VDR-deficient animals.22 By contrast, this “rescue diet” is unable to completely reverse growth plate abnormalities in 1α-hydroxylase–deficient mice, suggesting that 1,25(OH)2D3, acting through a receptor other than the classical VDR, may be essential for proper growth plate development.23 1,25(OH)2D3 has been shown to regulate the renin-angiotensin system; 1α-hydroxylase–deficient mice demonstrate cardiac hypertrophy and dysfunction, which are reversed with angiotensin-converting enzyme blockade.24,25 Thus, 1,25(OH)2D3 may be essential for cardiac health—a finding that could explain observational data suggesting that active vitamin D sterol therapy improves survival in patients treated with maintenance dialysis.26,27
Native 25-hydroxyvitamin D [25(OH)D] deficiency is prevalent in patients with CKD; low levels of this form of the hormone also contribute to altered mineral metabolism. Vitamin D may be made in the skin or ingested from the diet.28,29 Ultraviolet B (UVB) (290 to 315 nm) photons penetrate the skin and are absorbed by 7-dehydrocholesterol to form previtamin D3, which then spontaneously converts to vitamin D3. Vitamin D3 is extruded from the skin cell into the extracellular space, where it binds vitamin D–binding protein.30 Although vitamin D created in the skin is exclusively of the D3 form, dietary sources of vitamin D and food supplements may contain vitamin D2 (created through UV irradiation of ergosterol in yeast) or D3 (from animal sources, particularly fish). Vitamin D (both D2 and D3) undergoes hydroxylation by the liver, forming 25(OH)D.31 Subsequently, 25(OH)D is taken up by renal tubular cells through a megalin-dependent process and undergoes a second hydroxylation, facilitated by renal 1α-hydroxylase, to 1,25(OH)2D3, a more potent stimulator of gut calcium absorption.32,33 Conversion of 25(OH)D to 1,25(OH)2D3 is independent of 25(OH)D stores in the general population but becomes a substrate-dependent process in patients with CKD.34 Although the actions of 25(OH)D have been underemphasized in CKD, extrarenal 1α-hydroxylase activity may significantly contribute to 1,25(OH)2D3 production, even in anephric patients.35–37 Thus low levels of the precursor, 25(OH)D, exacerbate 1,25(OH)2 D3 deficiency in the context of CKD.
Levels of 25(OH)D below 32 ng/mL are associated with increased PTH levels, reduced bone mineral density (BMD),38 and increased rates of hip fracture39 in the general population and therefore represent insufficient vitamin D storage. It is interesting to note that 25(OH)D levels in the vast majority of the general population meet the definition of D insufficiency, and a large percentage—as many as 57% in one series of medical inpatients40—have serum levels less than 15 ng/mL, thus qualifying as vitamin D deficient. Prevalence is higher in individuals with darker skin pigmentation, with 52% of Hispanic and black adolescents from the same cohort meeting the criteria for vitamin D deficiency.41
Several recent studies have documented a high prevalence of 25(OH)D deficiency in patients with CKD.42,43 Patients with CKD are at increased risk for vitamin D deficiency for several reasons. Many are chronically ill with little outdoor (sunlight) exposure, and CKD dietary restrictions, particularly of dairy products, curtail the intake of vitamin D–rich food.44 When compared with individuals with normal kidney function, patients with CKD display decreased skin synthesis of vitamin D3 in response to sunlight.45 This is exacerbated in individuals with darker skin.46 Proteinuria also contributes to D deficiency in the CKD population; 25(OH)D, in combination with vitamin D–binding protein, is lost in the urine.47,48
Parathyroid Hormone
The human PTH gene is located on chromosome 11 and contains two introns, which separate three exons encoding the 5′ untranslated region (UTR), the prepro region and PTH, and the 3′ untranslated region. The initial translational product of the mRNA is prepro-PTH, a 115 amino acid single-chain polypeptide, which undergoes conversion to pro-PTH (90 amino acids) in the rough endoplasmic reticulum. Six additional residues are removed from the N-terminal of pro-PTH in the Golgi apparatus to form the biologically active 1-84 PTH. PTH then is stored in secretory granules prior to release into the bloodstream.49–51 Sustained increases in PTH secretion occur with progression of CKD. This prolonged stimulation leads to high turnover bone disease and to the development of parathyroid gland hyperplasia.
PTH levels increase in response to alterations in metabolism of calcium, 1,25(OH)2 D3, phosphorus, 25(OH)D, and FGF-23 (Fig. 69-2). With the progression of CKD, additional factors develop that sustain high production of PTH. Among these factors are alterations in the regulation of prepro-PTH gene transcription, post-transcriptional modifications of PTH mRNA, reductions in calcium-sensing receptor (CaSR) and vitamin D receptor expression in the parathyroids, autonomous activity of adenomatous parathyroid glands, and skeletal resistance to the calcemic actions of PTH.
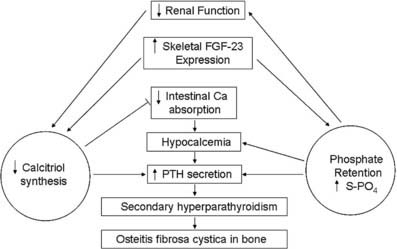
FIGURE 69-2. Pathogenesis of disordered mineral metabolism in chronic kidney disease (CKD). Dark arrows indicate effects of altered parathyroid hormone regulation; open arrows depict changes in fibroblast growth factor (FGF)-23 metabolism.
Calcium is the primary stimulus for PTH release. Calcium directly regulates PTH release via activation of the CaSR (vide infra). Activation of the CaSR decreases PTH release; deactivation increases PTH secretion. Serum calcium levels also regulate pre-pro PTH transcription via two negative calcium-response elements (nCaRE) located far upstream (−2.4 kbp and −3.5 kbp) of the PTH gene,52 and low serum calcium levels increase the stability of PTH mRNA by increasing levels of cytosolic parathyroid proteins, including AUF1,53 which binds to the 3′ UTR of the PTH mRNA. Thus, low levels of serum calcium present in CKD increase the amount of PTH release and the longevity of the PTH transcript. However, serum calcium levels are maintained within the normal range until late in the course of CKD, but serum PTH levels begin to rise much earlier, before any changes in serum calcium are evident.3 This suggests that factors other than calcium are important for the development of secondary hyperparathyroidism. Altered 1,25(OH)2 D3 metabolism may, in fact, be the initiating event early in the course of CKD.54–56
Apart from its effects on serum calcium levels, 1,25(OH)2D directly suppresses PTH levels. In conjunction with the VDR, 1,25(OH)2D3 binds negative vitamin D response elements in the promoter region of the PTH gene, thus inhibiting prepro-PTH gene transcription.57,58 In a positive-feedback loop, 1,25(OH)2D3 itself increases VDR gene expression in the parathyroid gland, further suppressing PTH gene transcription. 1,25(OH)2D3 also increases the expression of the CaSR, the expression of which is reduced in hyperplastic parathyroid tissues obtained from patients with secondary hyperparathyroidism.59 Vitamin D deficiency in animals is associated with decreased expression of CaSR mRNA in parathyroid tissue; 1,25(OH)2D3 therapy increases CaSR mRNA levels in a dose-dependent manner.60 Because 1,25(OH)2D3 is a potent inhibitor of cell proliferation, disturbances in renal calcitriol production and/or changes in VDR expression may be particularly important determinants of the degree of parathyroid hyperplasia and the extent of parathyroid gland enlargement in CKD.61
Phosphorous retention and hyperphosphatemia have been recognized for many years as important factors in the pathogenesis of secondary hyperparathyroidism. Phosphorous retention and hyperphosphatemia indirectly promote the secretion of PTH in several ways. Hyperphosphatemia lowers blood ionized calcium levels as free calcium ions complex with excess inorganic phosphate. The ensuing hypocalcemia stimulates PTH release. Increased phosphorus also impairs renal 1α-hydroxylase activity, which diminishes the conversion of 25(OH)D to 1,25(OH)2D3.16 Finally, phosphorus can directly enhance PTH synthesis. High serum phosphorous levels decrease cytosolic phospholipase A2 (normally increased by CaSR activation), leading to a decrease in arachidonic acid production with a subsequent increase in PTH secretion.62 Hypophosphatemia also decreases PTH mRNA transcript stability in vitro,63 suggesting that phosphorus itself affects serum PTH levels, probably by increasing the stability of the PTH mRNA transcript.
Although the actions of 25(OH)D have been underemphasized in dialyzed patients, evidence suggests that levels of this so-called “storage” form of vitamin D have both indirect and direct effects on PTH secretion. Extrarenal 1α-hydroxylase activity may contribute significantly to calcitriol production, even in anephric patients.35–37 Furthermore, recent evidence suggests the presence of 1α-hydroxylase in the parathyroid glands. 25(OH)D is converted inside the gland to 1,25(OH)2D, thereby suppressing PTH.64 25(OH)D administration suppresses PTH synthesis even when parathyroid gland 1α-hydroxylase is inhibited, indicating that 25(OH)D contributes to PTH suppression independent of its conversion to calcitriol.64 Indeed, recent studies have demonstrated that supplementation with ergocalciferol decreases serum PTH levels in patients with CKD.65,66 Such findings suggest that assessment of vitamin D status should be routinely performed in this patient population.67
Altered FGF-23 synthesis and secretion may contribute to increasing PTH levels through both indirect and direct mechanisms. Levels of FGF-23 rise as CKD progresses13,14 and contribute to declining 1,25(OH)2D levels. Lower 1,25(OH)2D levels, in turn, result in increasing PTH release. FGF-23 levels have been implicated in direct regulation of parathyroid gland function. In vitro and in vivo analysis of parathyroid glands from animals with normal renal function demonstrates that FGF-23 suppresses PTH secretion through a mechanism independent of its actions on vitamin D metabolism (vide infra).18,19
Alterations in parathyroid gland CaSR expression also occur in secondary hyperparathyroidism and may contribute to parathyroid gland hyperplasia. The CaSR is a seven-transmembrane G protein–coupled receptor with a large extracellular N-terminus, which binds acidic amino acids and divalent cations.68 Low extracellular calcium levels result in decreased calcium binding to the receptor, conformational relaxation of the receptor, and a resultant increase in PTH secretion,69 while activation of the receptor by high levels of serum calcium decreases PTH secretion.70,71 Expression of the CaSR is reduced by 30% to 70%, as judged by immunohistochemical methods in hyperplastic parathyroid tissue obtained from human subjects with renal failure.59,72 Decreased expression and activity of CaSR have been linked to decreased responsiveness in PTH secretion due to altered calcium levels.73 This decreased expression of the CaSR results in insensitivity to serum calcium levels with subsequent uncontrolled secretion of PTH. Increased stimulation of the CaSR by calcimimetics has been shown to decrease PTH cell proliferation, implicating the CaSR as a regulator of cell proliferation and PTH secretion.74
The link between the CaSR and vitamin D in cell cycling in the parathyroid gland is incompletely understood. However, some evidence suggests that vitamin D may decrease parathyroid hyperplasia by activating the CaSR. CaSR gene transcription is regulated by vitamin D through two distinct vitamin D response elements in the gene’s promoter region,75 suggesting that alterations in vitamin D metabolism in renal failure could account for changes in calcium sensing by the parathyroid glands, and that vitamin D may act upstream of the CaSR in preventing parathyroid cell hyperplasia.21
Once established, parathyroid enlargement is difficult to reverse because the rate of apoptosis in parathyroid glands is low, and the half-life of parathyroid cells is approximately 30 years.76 Chronic stimulation of parathyroid glands may lead to chromosomal changes that ultimately result in autonomous, unregulated growth and hormone release.77 Hyperplastic parathyroid tissue has been shown to develop inactivation of tumor suppressor genes MEN1, the retinoblastoma protein,78–80 and/or activating mutations of the RET proto-oncogene (MEN2a).81 Chromosomal translocations resulting in the parathyroid promoter driving cell cycle proteins (particularly cyclin D1) have been shown to be present in parathyroid adenomas.82 Even in the absence of somatic mutations, PTH secretion from enlarged parathyroid glands may become uncontrollable owing to the nonsuppressible component of PTH release from a large number of parathyroid cells. This alone may be sufficient to produce hypercalcemia and progressive bone disease in patients with end-stage renal disease.
Pathogenesis of Renal Bone Disease
ABNORMALITIES IN BONE TURNOVER, MINERALIZATION, AND VOLUME
Evaluation of skeletal histology by bone histomorphometry provides a method for understanding the pathophysiology of renal bone disease and a guide to its proper management. As recently recommended by the Kidney Disease Improving Global Outcomes (KDIGO) workgroup, three areas of bone histology are examined: bone turnover, mineralization, and volume, all of which may be altered in patients with CKD.1
Turnover
Traditionally, renal osteodystrophy has been classified primarily by alterations in bone turnover. Because PTH activates the PTH-related protein (PTHrP) receptor on osteocytes and osteoblasts, increasing cellular activity of both osteoblasts and osteoclasts,83,84 excessive levels of circulating PTH result in increased bone turnover (Figs. 69-3A and 69-3B).85 Serum PTH levels are inversely correlated with GFR, and most patients with GFR less than 50 mL/min have increased serum PTH levels and high turnover bone disease.86–88 The presence of CKD, however, markedly attenuates the effects of PTH on bone.89–90 Indeed, serum levels of PTH that are three to five times the normal range are associated with normal bone turnover in patients treated with maintenance dialysis, while similar PTH values in patients with mild to moderate kidney disease are associated with high turnover osteodystrophy.87,91,92 Although the precise mechanisms are poorly understood, uremia has been associated with this “skeletal resistance” to the actions of PTH. Uremic animals and humans display decreased PTH/PTHrP receptor mRNA expression in bone and growth plate.93,94 Hyperphosphatemia and alterations in vitamin D metabolism, among other factors, have been implicated in these changes, and calcitriol administration has been shown to partially restore the calcemic response to PTH in both experimental animals and patients with moderate CKD.95 Despite this “skeletal resistance” to PTH, many patients with end-stage kidney disease display bone biopsy evidence of PTH excess.
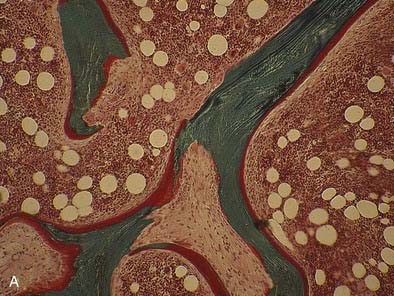
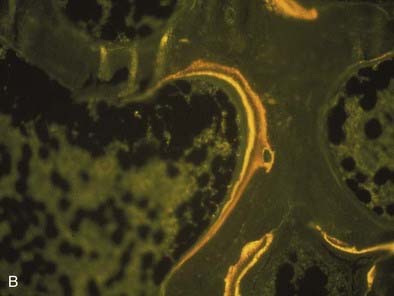
FIGURE 69-3. Bone histology, osteitis fibrosa. A, Under light microscopy, an increase in cellular activity, osteoid accumulation, erosion, and fibrosis are visible. B, An increase in double tetracycline labeling signifies an increase in bone turnover rate.
The bone in secondary hyperparathyroidism exhibits a marked increase in turnover with increased numbers of osteoblasts and osteoclasts and variable degrees of peritrabecular fibrosis. Activation of osteoclasts is mediated through PTH96–99; the result is increased resorption of both mineral and matrix along the trabecular surface and within the haversian canals of cortical bone.100 Such increased cellular activity can occur secondary to a nonspecific reaction to local factors, such as insulin-like growth factor-1 (IGF-1), cytokines, or fracture, or as the result of systemic stimuli, such as increased thyroxine or PTH. One characteristic of high bone turnover is increased quantities of woven osteoid, exhibiting haphazard arrangements of collagen fibers in contrast to the usual lamellar pattern of osteoid in normal bone. Woven osteoid can become mineralized in patients with advanced kidney disease in the absence of vitamin D; however, the calcium may be deposited as amorphous calcium phosphate rather than hydroxyapatite.101
At the other end of the spectrum of bone turnover, adynamic bone disease is characterized by normal osteoid volume, an absence of fibrosis, and a reduced bone formation rate, as indicated by a reduced or absent double tetracycline label on bone histomorphometry (Fig. 69-4).102,103 A paucity of osteoblasts and osteoclasts is observed.85 The histologic features of adynamic renal osteodystrophy, in the absence of aluminum deposition in bone, cannot be distinguished from the histologic features of corticosteroid-induced osteoporosis or age-related or postmenopausal osteoporosis. Therefore, it is not possible to determine whether osteoporosis accounts for decreases in osteoblastic activity and bone formation in patients with adynamic renal osteodystrophy unless the amount of trabecular bone is reduced. Decreases in bone mass and histologic evidence of trabecular bone loss are not integral features of the adynamic lesion of renal osteodystrophy when other causes of osteoporosis can be excluded. Adynamic bone is associated with low PTH levels, low alkaline phosphatase levels, high serum calcium levels, and a propensity for increased vascular calcification.104,105
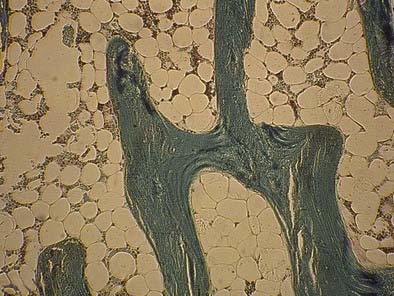
FIGURE 69-4. Bone histology, adynamic bone. Under light microscopy, decreased cellular activity occurs with minimal osteoid accumulation.
In the 1970s and 1980s, aluminum intoxication was largely responsible for the development of adynamic bone and osteomalacia in patients with CKD. Two distinct patterns of aluminum intoxication were identified: (1) absorbed from the aluminum content of water used to prepare dialysate solution,106–108 and (2) intestinal aluminum absorption after ingestion of large doses of aluminum hydroxide.109–115 The neurologic syndrome of “dialysis encephalopathy” and a bone disease manifested by fractures, pain, persistent hypercalcemia, and osteomalacia were the main clinical features. Although the prevalence of aluminum bone disease in developed countries is now very low, the prevalence of adynamic renal osteodystrophy not associated with aluminum intoxication has increased substantially over the past few years in adult patients receiving regular dialysis.116 Currently, adynamic renal osteodystrophy is commonly associated with disorders such as age-related or postmenopausal osteoporosis, steroid-induced osteoporosis, hypoparathyroidism (idiopathic or surgically induced), and diabetes mellitus, and with overtreatment with calcium and vitamin D therapy.117
Because PTH is the major determinant of bone formation and skeletal remodeling in renal failure, oversuppression of PTH secretion can result in adynamic renal osteodystrophy. Approximately 40% of those treated with hemodialysis and more than half of adult patients undergoing peritoneal dialysis have serum PTH levels that are only minimally elevated or that fall within the normal range; such values are typically associated with normal or reduced rates of bone formation and turnover.103 Prolonged treatment with calcium-containing phosphate-binding medications and the use of high-dialysate calcium concentrations also contribute to low bone turnover.118 Calcitriol may directly suppress osteoblastic activity when given intermittently in large doses to patients receiving regular dialysis.119
The long-term consequences of adynamic renal osteodystrophy not attributable to aluminum toxicity remain to be determined, but concerns have been raised about increases in the risk for skeletal fracture and delayed fracture healing due to low rates of bone remodeling.119 The development of soft tissue and vascular calcifications has been associated with adynamic bone disease in cross-sectional studies.105 In prepubertal children, adynamic renal osteodystrophy has been associated with a reduction in linear growth.117
Mineralization
Although renal osteodystrophy has traditionally been defined by lesions in bone turnover, alterations in skeletal mineralization are also prevalent in CKD.92 Increases in unmineralized bone (osteoid) in conjunction with delayed rates of mineral deposition are common.92,100 Defective mineralization associated with low to normal bone turnover is termed osteomalacia.1 Histomorphometric characteristics of osteomalacia include the presence of wide osteoid seams, an increased number of osteoid lamellae, an increase in the trabecular surface covered with osteoid, and a diminished rate of mineralization or bone formation, as assessed by double tetracycline labeling. Fibrosis is often absent.85 In patients on long-term dialysis, osteomalacia that is refractory to vitamin D therapy is most commonly a result of aluminum intoxication.120
Although the mechanisms of skeletal mineralization are incompletely understood, factors such as 25(OH)D deficiency and altered FGF-23 metabolism have been implicated in their pathogenesis. In the general population, nutritional 25(OH)D deficiency results in osteomalacia, and a similar phenotype may occur in patients with CKD. FGF-23 may play a role; both overexpression121–123 and ablation of FGF-23124,125 in mice with normal renal function are associated with abnormal mineralization of osteoid, although by different mechanisms. The phosphaturic effects of increased FGF-23 may cause rickets and osteomalacia through an insufficiency of mineral substrate. The mechanisms leading to impaired mineralization in FGF-23–null animals, which have severe hyperphosphatemia and normal or elevated serum calcium levels, remain uncertain; however, osteomalacia in these animals suggests that FGF-23 may play a direct role in skeletal mineral deposition. Although the ramifications of defective mineralization remain to be established, increased fracture rates and bone deformities are prevalent in patients with CKD despite adequate control of bone turnover. These complications may be due, in part, to alterations in bone mineralization.
Treatment with anticonvulsant therapy may contribute to the development of osteomalacia in patients with kidney disease. Long-term ingestion of phenytoin and/or phenobarbital is associated with a high incidence of osteomalacia in nonuremic patients.126 These findings may be due in part to alterations in vitamin D metabolism.127
Osteitis fibrosa cystica, a finding of secondary hyperparathyroidism, can coexist with defective mineralization in some patients; this pattern is called a “mixed” lesion.128 Patients with mixed lesions often display high serum PTH and alkaline phosphatase levels, along with lower serum calcium levels. Mixed lesions are seen with high turnover bone disease in patients who are developing aluminum toxicity, or in patients with low turnover aluminum-related bone disease during desferoxamine (DFO) therapy (vide infra).129 In these cases, mixed lesions represent a transitional stage between high turnover and low turnover bone disease.
Volume
Because PTH is anabolic at the level of trabecular bone, high levels of serum PTH are typically associated with increases in bone volume, trabecular volume, and trabecular width.92,116,130,131 However, bone volume may also be low (termed osteoporosis), particularly in individuals with underlying age-related bone loss and in those treated with corticosteroids. Osteoporosis in the general population is associated with increased risk for hip fractures and mortality.132 Thus, bone volume is considered a critically important parameter of bone histology. The impact of osteoporosis on morbidity and mortality in the CKD population, however, remains to be defined.
Clinical Manifestations
The symptoms and signs of renal osteodystrophy are usually nonspecific, and laboratory and radiographic abnormalities generally pre-date clinical manifestations. Some specific symptoms and syndromes do occur, however.
BONE PAIN
Bone pain is a common manifestation of severe bone disease in patients with advanced kidney disease. It usually is insidious in appearance and often is aggravated by weight bearing or a change in posture. Physical findings are often absent. Pain is most common in the lower part of the back, hips, and legs but may occur in the peripheral skeleton. Occasionally, sudden appearance of pain around the knee, ankle, or heel can suggest acute arthritis; such pain is not usually relieved by massage or local heat. Bone pain is more common and often is more marked in patients with aluminum-related bone disease than in those with osteitis fibrosa cystica, but marked variability may be seen from one patient to another.133
In patients on long-term dialysis, carpal tunnel syndrome and chronic arthralgias often occur in association with the deposition of β2-microglobulin amyloid in articular and periarticular structures.134 Arthralgias usually are bilateral and most commonly affect the shoulders, knees, wrists, and small joints of the hand; symptoms typically are worse with inactivity and at night.135,136
MUSCLE WEAKNESS
Proximal myopathy can be marked in patients with advanced kidney disease. Symptoms appear slowly. Patients may note difficulty climbing stairs or rising from a low chair, or they may have difficulty raising their arms to comb their hair. This proximal muscle weakness resembles that found in 25(OH)D deficiency and in primary hyperparathyroidism. Plasma levels of muscle enzymes usually are normal, and electromyographic changes are nonspecific.
The pathogenesis of this myopathy is not clear, and several different mechanisms, including secondary hyperparathyroidism, phosphate depletion,137 abnormal vitamin D metabolism, and aluminum intoxication, have been implicated.138 Improvement in gait posture has been reported in children with moderate renal failure after treatment with 1,25(OH)2D3, and muscle weakness improves rapidly in affected adult patients with end-stage kidney disease.139 Improvement in muscular strength has been observed after treatment with 25(OH)D3, after subtotal parathyroidectomy, after successful renal transplantation, and after chelation therapy with deferoxamine for aluminum intoxication.
SKELETAL DEFORMITIES
Bone deformities are common in uremic children because their bones undergo growth, modeling, and remodeling. In adult patients, skeletal deformities also arise from abnormal remodeling or recurrent fractures.140 In children, bone deformities of the femora and wrists arise from slipped epiphyses.141 This problem is most common during the preadolescent period and is most frequent in patients with long-standing congenital kidney disease. In adults with kidney disease, particularly those with aluminum-related bone disease, skeletal deformities may be characterized by lumbar scoliosis, kyphosis, and deformities of the thoracic cage.140
GROWTH RETARDATION
Growth retardation is the hallmark of CKD in children. Protein and calorie malnutrition, metabolic acidosis, end-organ growth hormone resistance, and renal bone disease are most commonly implicated in growth failure.142 Despite correction of acidosis and anemia, normalization of serum calcium and phosphorous levels, and vitamin D sterol therapy replacement, most children with CKD continue to grow poorly. Growth failure worsens as renal function declines; the average height of children with even mild CKD (GFR 50 to 70 mL/minute/1.73 m2) is 1 standard deviation (SD) below the average for healthy children. Moderate CKD (GFR 25 to 49 mL/minute/1.73 m2) is associated with a height SD of −1.5, and, at the time of initiation of dialysis, the mean height SD is −1.8. Boys, younger patients, and those with prior renal transplants are at greatest risk for growth failure.143
Acidosis has been linked to delayed linear growth in patients with renal tubular acidosis and normal renal function, and correction of metabolic acidosis often leads to acceleration in growth velocity.144 Acidotic rats have been found to have decreased growth hormone (GH) secretion, serum insulin-like growth factor 1 (IGF-1), and hepatic IGF-1 mRNA expression. Moreover, metabolic acidosis has been shown to inhibit the effects of GH in rats with normal and decreased renal function.145–147 Growth plate mRNA levels of GH receptor, IGF-1 receptor, and IGF-1 expression are downregulated, and IGF-binding proteins are upregulated.148 In adults treated with maintenance dialysis, correction of acidosis has been shown to decrease the progression of secondary hyperparathyroidism and to improve skeletal mineralization.149
Calcitriol deficiency has been thought to contribute to growth retardation and bone disease in children with CKD. Secondary hyperparathyroidism remains prevalent in children with advanced renal disease, and osteitis fibrosa continues to be the most common skeletal lesion of renal osteodystrophy in those undergoing regular dialysis despite treatment with daily doses of oral calcitriol.116,150 Secondary hyperparathyroidism contributes to growth retardation, although optimal target values for PTH in children at all stages of CKD remain controversial. In children with moderate CKD, some data indicate that normal growth velocity is achieved when PTH levels are maintained within the normal range151; others have demonstrated a linear correlation between growth and PTH levels in the same patient population, with those with the highest PTH values displaying the highest growth velocity.152 Treatment for secondary hyperparathyroidism with large, intermittent doses of calcitriol and calcium-based phosphate binders has been shown to significantly reduce bone formation and suppress osteoblastic activity in both adults and children.95,153 However, adynamic bone disease may develop and linear bone growth decrease, despite serum PTH levels in the K/DOQI117,154 recommended range during intermittent vitamin D sterol therapy. Maintaining serum PTH levels at between 300 and 500 pg/mL reduces the frequency of these complications.131 The mechanisms by which calcitriol inhibits epiphyseal growth plate cartilage remain poorly understood. However, it is well known that calcitriol exerts dose-dependent inhibitory effects on cell proliferation of chondrocytes and osteoblasts in vitro. In addition, vitamin D sterols increase expression of a number of IGF-binding proteins (IGFBPs). IGFBP-2, -3, -4, and -5 sequester IGF-1 and may exert IGF-1–independent antiproliferative effects through their own receptors.155–159
GH resistance also contributes to impaired linear growth in renal failure. In CKD, poor growth develops despite normal or increased serum GH levels.160,161 Uremia has been associated with diminished hepatic GH receptor and IGF-1 mRNA expression, defects in postreceptor GH-mediated signal transduction,162,163 reductions in serum GH-binding protein levels,164 and increased synthesis and reduced clearance of IGF binding proteins.138,164,165 Improved growth velocity during recombinant human GH (rhGH) therapy has been ascribed to increased bioavailability of IGF-1 to target tissues. Children who are treated with maintenance dialysis respond less well to rhGH therapy than do children with less severe CKD, but the mechanisms for differences in response to GH therapy remain to be determined.
EXTRASKELETAL CALCIFICATION
Extraskeletal calcification has been associated with uremia for many years166 and has been included recently in the definition of CKD-MBD.1 Vascular calcifications are associated with increased mortality. These lesions have their origins in CKD prior to dialysis and begin in childhood (Fig. 69-5).167,169 The mortality rate in adults and children with CKD is markedly higher than that in the general population, and cardiovascular disease is the leading cause of death in both children and adults treated with maintenance dialysis.168,170 In contrast to the calcified atherosclerotic plaques that develop in the vascular intima of aging individuals with normal kidney function, uremia facilitates calcification of the tunica media. This form of calcification is associated with decreased distensibility of blood vessels, causing a rigid “lead pipe” pathology that is associated with increased risk for congestive heart failure.1 Electron beam computed tomography (EBCT) is used in assessment of vascular calcifications in the adult population, and measurements in young adults who were treated with maintenance dialysis as children demonstrated that a significant proportion of this population has evidence of vascular calcification.168 Carotid ultrasound measurement of intima medial thickness (IMT) has been validated for the assessment of cardiovascular pathology in children, with increased thickness associated with worsening disease.169,171
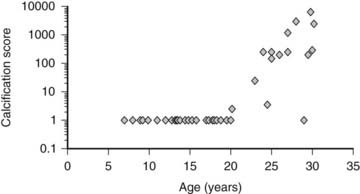
FIGURE 69-5. Coronary artery calcification scores in 39 children and young adults with end-stage renal disease who were treated by dialysis, according to age. Coronary artery calcification was assessed by electron beam computed tomography. The scale on the y-axis is logarithmic.
(Data from Goodman WG, et al: Coronary artery calcification in young adults with end-stage renal disease who are undergoing dialysis, N Engl J Med 106:100–105, 2000.)
Hypercalcemia, hyperphosphatemia, elevated levels of the calcium x phosphorous product, and high doses of vitamin D sterols167,168 all have been implicated in the pathogenesis of cardiovascular calcification. However, 40% of adult patients with stage 3 CKD, without these risk factors, show evidence of calcification,172 suggesting a role for uremic factors other than high levels of calcium and phosphorus. Indeed, vascular tissues in the uremic milieu express osteoblast differentiation factors.173 Osteoblasts and vascular smooth muscle cells have a common mesenchymal origin; core binding factor-1 (Cbfa1) is thought to trigger mesenchymal cell-to-osteoblast transformation. Mice that are deficient in Cbfa1 fail to mineralize bone,174 and arteries obtained from patients undergoing renal transplantation show increased levels of this protein.173 Upregulation of the sodium-dependent phosphate transporter PIT-1 likely also contributes to increased calcification,175 and upregulation of pro-mineralization factors such as osteopontin, bone sialoprotein, osteonectin, alkaline phosphatase, type I collagen, and bone morphogenic protein-2 (BMP-2) is potentiated by the uremic milieu.176–179 By contrast, expression of calcification inhibitors such as fetuin A, matrix gla protein, and klotho is suppressed.180–183 Levels of circulating FGF-23 also may contribute, as high levels of the protein are associated with increased mortality in adult dialysis patients.184 Klotho, the cofactor necessary for the actions of FGF-23, has been implicated in vascular calcification. Animals lacking the Klotho gene display elevated levels of calcium, phosphorus, and vitamin D, along with vascular calcification and premature aging. CKD is associated with low circulating levels of Klotho,183 which has been implicated in the regulation of sodium/phosphate co-transport in the aorta.185
Because the pathophysiology of cardiovascular disease in CKD is multifactorial, treatment strategies are also multifaceted and vary according to stage of CKD. Therapies that are effective in early CKD may not be effective in later stages—lipid-lowering agents decrease mortality in adults with CKD186 and in those with stable renal allografts187 but have not been shown to benefit patients treated with dialysis.188 By contrast, normalization of mineral metabolism (attained by avoiding hypercalcemia and hyperphosphatemia, limiting calcium intake, and avoiding adynamic bone) is effective at slowing the progression of cardiovascular calcification in patients treated with maintenance dialysis.154,168,189,190 Thus, at different stages of CKD, the relative importance of individual risk factors and the value of different biomarkers may vary.
CALCIPHYLAXIS
This unique syndrome, which is characterized by ischemic necrosis of the skin, subcutaneous fat, and muscles, can develop in patients with advanced renal failure not yet treated by dialysis, in those treated with regular dialysis, and in patients with well-functioning kidney transplants.191 The pathogenesis of this syndrome is uncertain. Extensive medial calcifications of medium-sized arteries are noted; such calcifications commonly exist in patients with renal disease without causing gangrene or ulcerations, and it is not clear that vascular calcifications per se are the cause of the ischemic necrosis. Two distinct types of the syndrome are recognized: proximal calciphylaxis, which affects the thighs, abdomen, and chest wall, and acral calciphylaxis, which involves sites distal to the knees and elbows, such as the toes, fingers, and ankles.192 The former has a terrible prognosis, with death occurring in more than 80% to 90% of affected patients. This syndrome is often accompanied by morbid obesity and hypoalbuminemia. Many patients have severe secondary hyperparathyroidism, and most have a history of severe and uncontrolled hyperphosphatemia.192 Some patients may have defective regulation of coagulation.193 The appearance of this syndrome in renal transplant recipients receiving glucocorticoids suggests that steroids may also play a role. Patients with calciphylaxis frequently die of secondary infection.
A significant number of patients improve after parathyroidectomy, and a few have healed after substantial reductions in levels of serum phosphorus. Parathyroidectomy and aggressive control of serum phosphate levels therefore are indicated in those with evidence of severe secondary hyperparathyroidism. However, although ischemic lesions and medial vascular calcifications are common in uremic patients with diabetes, such lesions rarely improve after parathyroidectomy. Thus, parathyroid surgery should be reserved for those diabetic patients with calciphylaxis who have clear evidence of severe secondary hyperparathyroidism. Calcimimetics,194 as well as sodium-thiosulfate (a calcium chelating agent) and pamidronate,195 have been used effectively in some individuals. Hyperbaric oxygen therapy also has been advocated.195
DIALYSIS-RELATED AMYLOIDOSIS
Several clinical syndromes that arise as a consequence of dialysis-related amyloidosis can mimic the clinical features of renal osteodystrophy, or amyloidosis can occur concurrently with uremic bone disease. Dialysis-related amyloidosis arises from the deposition in bone and periarticular structures of a specific type of amyloid composed of β2-microglobulin.196 In addition to β2-microglobulin, the amyloid deposits contain advanced glycosylation end products, which may account for their uptake by certain collagen-rich structures.197,198 The frequency of its clinical manifestation rises markedly in patients treated with regular dialysis for longer than 5 to 10 years, and it is much more common in patients who start dialysis when older than 50 years.199 Blood levels of β2-microglobulin are strikingly elevated in all patients with end-stage renal disease because of failure of normal renal clearance and catabolism. Clinical manifestations include (1) carpal tunnel syndrome; (2) destructive or erosive arthropathy involving the large and medium-sized joints, with shoulder, knee, hip, or back pain being common manifestations; (3) spondyloarthropathy, most commonly affecting the cervical spine; and (4) subchondral, thin-walled cysts of bone, most commonly affecting the carpal bones, the humoral and femoral heads, the distal end of the radius, the acetabulum, and the tibial plateau. These subchondral cysts at times are confused with brown tumors of secondary hyperparathyroidism, although their location and occurrence in multiple sites make them quite different from brown tumors. Nonetheless, in a patient who has undergone long-term dialysis, this syndrome may be a potential cause of neuromuscular and periarticular symptoms usually considered to be due to secondary hyperparathyroidism or aluminum accumulation. Parathyroid surgery should be avoided in dialysis patients whose severe musculoskeletal symptoms have not improved with calcitriol therapy; symptoms in these patients actually may be due to dialysis-related amyloidosis. Specific diagnosis of the latter is made from the biopsy demonstration of amyloid composed of β2-microglobulin; however, the diagnosis can be strongly suspected from the clinical features, the presence of multiple thin-walled cysts, or the demonstration in periarticular sites of presumed amyloid tissue on ultrasonography.200 Its management is difficult and largely unsatisfactory, but successful renal transplantation leads to rapid disappearance of symptoms and no further progression of the bone lesions on radiographs.201 In patients who remain on maintenance dialysis, the use of high-flux dialyzer membranes may decrease amyloid accumulation202; the long-term effects of this form of therapy on symptoms remain to be determined.
Diagnostic Evaluations
BIOCHEMICAL DETERMINATIONS
Phosphorus
Hyperphosphatemia has been found to be an independent risk factor for mortality in patients treated with maintenance dialysis.203 Hemodialysis and continuous ambulatory peritoneal dialysis (CAPD) remove phosphate, but dietary phosphate restriction and the use of phosphate-binding agents are required by 90% to 95% of patients undergoing treatment with dialysis. To prevent the development and progression of secondary hyperparathyroidism and vascular calcification, such measures as dietary phosphate restriction and the use of phosphate-binding medications should be initiated to maintain serum phosphorous levels within age-appropriate levels and calcium phosphorous products lower than 55 mg2/dL.67,204
Calcium
Hypocalcemia often resolves during treatment with calcium-containing phosphate binders, with vitamin D, and with initiation of dialysis. The development of hypercalcemia in patients undergoing regular dialysis warrants prompt and thorough investigation. Conditions associated with hypercalcemia include marked hyperplasia of the parathyroid glands as a result of severe secondary hyperparathyroidism, aluminum-related bone disease, therapy with calcitriol or other vitamin D sterols, administration of large doses of calcium carbonate or other calcium-containing compounds, immobilization, malignancy, and granulomatous disorders, such as sarcoidosis or tuberculosis in which extrarenal production of 1,25(OH)D occurs.102,205,206 Basal serum calcium levels are also higher in patients with adynamic bone than in subjects with other lesions of renal osteodystrophy, and episodes of hypercalcemia are common.92 Because skeletal calcium uptake is limited in adynamic lesions, calcium entering the extracellular fluid from dialysate or after intestinal absorption cannot be buffered adequately in bone, and serum calcium levels rise.207 Maintaining total calcium intake at between 1500 and 2000 mg per day, the use of new-generation vitamin D sterols, and treatment with calcimimetic agents help to limit episodes of hypercalcemia.
Magnesium
The net intestinal absorption of magnesium generally is normal or only very slightly reduced in patients with renal failure,204 yet serum magnesium levels often increase with advanced kidney disease as the result of reduced renal excretion. During hemodialysis, serum magnesium levels generally are increased if the dialysate magnesium concentration is maintained at 1.75 mEq/L; however, magnesium levels remain within the upper range of normal with dialysate magnesium concentrations of 0.5 mEq/L. The use of magnesium-containing laxatives or antacids should be avoided as these may cause an abrupt increase in serum magnesium levels in patients with kidney disease.208 Serum magnesium levels should be measured frequently and regularly if magnesium-containing medications are used. Rarely, hypomagnesemia can develop in uremic patients with severe malabsorption or diarrhea.204
Alkaline Phosphatase
Serum alkaline phosphatase values are fair markers of the severity of secondary hyperparathyroidism in patients with renal failure. Osteoblasts normally express large amounts of the bone isoenzyme of alkaline phosphatase, and serum levels usually are elevated when osteoblastic activity and bone formation rates are increased. High levels generally reflect the extent of histologic change in patients with high turnover lesions of renal osteodystrophy, and values frequently correlate with serum PTH levels.209 Serum total alkaline phosphatase measurements are also useful for monitoring the skeletal response to treatment with vitamin D sterols in patients with osteitis fibrosa; values that decrease over several months usually indicate histologic improvement.117 Bone-specific alkaline phosphatase activity may be useful in predicting the histologic lesions of renal osteodystrophy, but whether these values are superior to total alkaline phosphatase levels remains to be demonstrated. Serum alkaline phosphatase levels may increase early in the course of treatment for aluminum-related bone disease with the chelating agent deferoxamine. Levels also may increase during therapy with recombinant human growth hormone in pediatric patients with renal failure.210
Assays for bone-specific alkaline phosphatase and measurements of serum osteocalcin levels provide additional information about the level of osteoblastic activity in patients with CKD.211 Osteocalcin levels generally are elevated in renal failure, but values may help distinguish between patients with high turnover and those with low turnover skeletal lesions.211,212 If these assays are not available, measurement of the heat-stabile and heat-labile fractions of alkaline phosphatase helps to separate skeletal from hepatic causes of elevated total alkaline phosphatase levels.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


