This chapter focuses on Hashimoto’s thyroiditis, also known as chronic lymphocytic thyroiditis, chronic autoimmune thyroiditis, and lymphadenoid goiter. In the early stage, patients are euthyroid and have no or a very small goiter. Chronic thyroiditis is subclinical, and the only evidence of autoimmune thyroiditis is the presence of antithyroid antibodies in the serum. Postmortem histologic examination has revealed that positive tests for serum antithyroid antibodies, especially antithyroid microsomal antibodies (now known as thyroid peroxidase antibodies [TPOAb]), in subjects without overt thyroid disease indicate the presence of lymphocytic infiltration into the thyroid.5 As the disease progresses, patients show a firm, diffuse goiter of small to moderate size and generally are said to have chronic autoimmune thyroiditis. Their thyroid function is variable, ranging from euthyroidism to thyrotoxicosis. A large, firm goiter develops when the disease is more advanced; this type is the classical or goitrous Hashimoto’s disease. Further progression of the immune process results eventually in atrophic thyroiditis in association with hypothyroidism. This combination represents the final stage of Hashimoto’s disease.
In the general population, the percentages of women and men with serum TPOAb and antithyroglobulin antibodies (TgAb) increase with age, from about 10% in women in reproductive age to as many as 19% or more among elderly women.6 The prevalence in men is much less—about 5%. Subclinical autoimmune thyroiditis is further evidenced by the fact that thyroid dysfunction develops after delivery in up to 50% of women with positive TPOAb measured in early pregnancy.7 Therefore, when subclinical autoimmune thyroiditis is included, chronic thyroiditis is a very common disease. One in 10 to 30 women in the general population has autoimmune thyroiditis.
Pathology
In the classic form of Hashimoto’s thyroiditis (struma lymphomatosa) with a firm, enlarged thyroid, the normal follicular structure is extensively replaced by lymphocytic and plasma cell infiltrates with the formation of lymphoid germinal centers (Fig. 83-1). Thyroid follicles remain isolated or in small clusters, are small or atrophic, and are empty or contain sparse colloid. Some persistent follicular epithelial cells are transformed into Askanazy cells, which have an eosinophilic granular cytoplasm. These cells are found in many other thyroid diseases and probably represent a damaged state of epithelial cells. Fibrosis of variable extent and lymphocytic infiltration are found in the interstitial tissue.
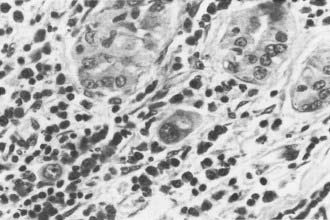
FIGURE 83-1. Hashimoto’s thyroiditis. Note atrophic follicles, absent colloid, and infiltrate of lymphocytes, plasma cells, and immunoblasts (lower left). (Hematoxylin and eosin [H&E], ×200)
(From Livolsi VA. In Falk SA, ed: Thyroid disease: endocrinology, surgery, nuclear medicine and radiotherapy, New York, 1990, Raven Press.)
Autoimmune Abnormalities
INITIATION OF THYROID AUTOIMMUNITY
Similar to other autoimmune diseases, autoimmune thyroiditis can arise from a breakdown of self-tolerance to thyroid antigens. Immunologic self-tolerance is thought to be induced during the perinatal period, when immature lymphocytes are exposed to self-antigens.8 At this critical point, clonal deletion or induced anergy of autoreactive T cells in the thymus provides self-tolerance to autoantigens. If an abnormality occurs during this period, self-tolerance might not be induced,8 and autoimmune thyroiditis might develop. An early theory was that a genetically induced organ-specific suppressor T lymphocyte defect could deregulate a thyroid-specific helper T cell population.9 This view has required redefinition in relation to the role of regulatory T cells in thyroid autoimmunity.10 Further breakdown in self-tolerance may be induced by altered self-antigen, exposure to environmental antigens that mimic a self-antigen, polyclonal immune activation, or idiotype cross-reaction of self-antigens. These factors may augment low levels of autoimmune thyroiditis. For example, infection, drugs, or other factors might activate autoreactive helper lymphocytes. Locally produced interferon-γ (IFN-γ) may induce major histocompatibility complex (MHC) class II antigen expression on thyroid cell surfaces, which may promote autoimmunity.11 Environmental factors also play an important role in the pathogenesis of autoimmune thyroiditis. A summary of the genetic and environmental factors that predispose to the condition is shown in Table 83-2. Details are described in subsequent sections.
Table 83-2. Factors Predisposing to Autimmune Thyroiditis
| Genetic | Environmental |
|---|---|
| HLA | Smoking |
| CTLA-4 | Stress |
| PTPN22 | Iodine and selenium |
| TG | Drugs (amiodarone, lithium, interleukin-2, interferon-α, HAART, GM-CSF) |
| Irradiation, infection | |
| Pregnancy and post partum |
GM-CSF, Granulocyte-macrophage colony-stimulating factor; HAART, highly active antiretroviral therapy.
ENVIRONMENT AND THYROID AUTOIMMUNITY
Three studies all imply that smoking may in fact protect against development of TPO antibodies12 and inferentially against chronic lymphocytic thyroiditis. The mechanism for these findings is not clear, although it is known that smoking is a definite risk factor for the development of Graves’ ophthalmopathy and, to a lesser extent, Graves’ disease. Although stress is thought to contribute to the onset of Graves’ hyperthyroidism, no good data relate stressful life events to involvement in the origin of Hashimoto’s disease. Iodine administration is known to induce thyroiditis in susceptible animals by affecting the antigenicity of thyroglobulin.13,14 In contrast to iodine, it is a deficiency of selenium that has been noted to cause increased thyroid volume and reduced echogenicity, together with reduction in immune competence.15 Amiodarone contains 37% iodine, and many of its effects on the thyroid are iodine mediated; in addition, amiodarone administration results in the appearance of thyroid antibodies in patients who have preexisting thyroid autoimmunity.16 A similar situation occurs in patients who are receiving lithium therapy.17 Both of these drugs may have specific immunomodulatory effects, which exacerbate thyroid autoimmunity in Hashimoto’s thyroiditis. Interleukin (IL)-2 and Interferon-α (IFN-α) both cause changes in the immune system characterized by alterations in lymphocyte subsets, which may result in autoimmune thyroid disease.18 Highly active antiretroviral therapy (HAART) and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been noted to be associated with small increases in thyroid autoimmunity in some studies.19,20 External irradiation, as an accident (e.g., Chernobyl), can result in the expression of autoimmune thyroid disease and the emergence of thyroid antibodies.21 Although infection (viral or bacterial) is an attractive factor to be considered as a cause of autoimmune thyroid disease, the data related to Hashimoto’s thyroiditis are tentative at best.22,23 The molecular mimicry hypothesis regarding the increased incidence of Yersinia enterocolitica noted in patients with Graves’ disease has not been confirmed as a causative factor, and this serology is not seen in Hashimoto’s disease. The switch in peripheral lymphocyte pattern from Th2 during pregnancy to a Th1 state post partum is associated with the so called “immune rebound,” which is characterized by a rapid rise in titers of thyroid antibodies in women who are known to have these antibodies in early gestation. In about 25% to 30% of these women, permanent autoimmune hypothyroidism occurs. Transient postpartum thyroiditis is seen in the remaining antibody-positive women. Thus pregnancy must be regarded as a specific cause of autoimmune thyroid disease in predisposed women. From the foregoing, it will be appreciated that although environmental factors are important, it is necessary to impose these on the appropriate genetic background to initiate the immune process. This, together with an overview of the immune abnormalities seen in Hashimoto’s disease, will be discussed in the following sections.
Genetic Factors
It is widely known that autoimmune thyroid diseases (both Hashimoto’s thyroiditis and Graves’ disease) occur in families.24 Fig. 83-2 shows the age at diagnosis in 400 patients with Hashimoto’s disease (panel A) and shows that those with a positive family history present at a younger age than those who have no family history (panel B). This tendency could be due to genetic predisposition, as well as to environmental influences. Studies of genetic predisposition have revealed that autoimmune thyroid diseases are often associated with particular genetic markers. These markers include histocompatibility lymphocytic antigens, allotypes of immunoglobulin heavy chains, and variations in the T cell receptor (TCR) and the thyroid peroxidase (TPO). More recently, the association of Hashimoto’s disease with variants of CTLA-4, PTNP22, and thyroglobulin has been documented.25 These associations have been examined by linkage analysis, association analysis, and whole genome screening. Findings described in reports are not always consistent with each other, probably because of the subjects chosen and the small sizes of some studies. The main susceptibility genes are shown in Table 83-3.
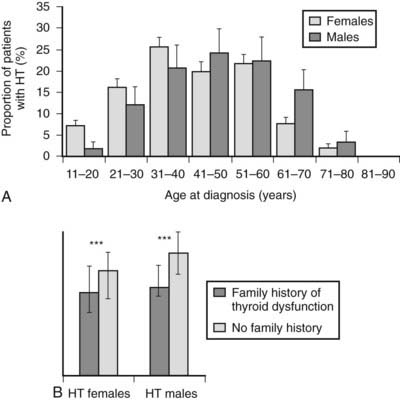
FIGURE 83-2. Age at diagnosis of Hashimoto’s thyroiditis (A) and median age at diagnosis of those with and without a family history of thyroid dysfunction (B).
(From Manji N, Carr-Smith JD, Boelaert K, et al: Influences of age, gender, smoking, and family history on autoimmune thyroid disease phenotype, J Clin Endocr Metab 91:4873–4880, 2006.)
Table 83-3. Susceptibility Genes for Hashimoto’s Thyroiditis
| Gene | Associated Variants | Population Association |
|---|---|---|
| HLA-DR | DR3,DR5 (goitrous) | Caucasian |
| DR3 and HLA B8 (atrophic) | Caucasian | |
| DR9 and HLA-Bw46,87 | Chinese | |
| HLA-DQ | DQw2 (link dis HLA DR3) | Cauasian |
| DQ A0301 (link dis HLA DR4) | Caucasian | |
| DQ B0201 (link dis HLA DR3) | Caucasian | |
| CTLA-4 | A/G49SNP, CT60 SNP | Caucasian, Japanese |
| 3′ UTR AT microsatellite | Koreans, Chinese | |
| PTPN22 | R620W SNP | Caucasian |
| Thyroglobulin | S734A SNP | Caucasian |
| T2334C SNP | Japanese | |
| M1028V, R1999W SNP |
Link dis, Linkage disequilibrium; SNP, single-nucleotide polymorphism.
Data from Jacobson EM, Tomer Y: The genetic basis of thyroid autoimmunity, Thyroid 17:949961, 2007.
Recent studies have highlighted the possibility of association of vitamin D receptor gene polymorphisms with increased risk for Hashimoto’s disease among the Chinese26 and possibly also in the Croatian population.27 Other possible candidate susceptibility genes for Hashimoto’s include an IL-6 gene promoter polymorphism28 and a polymorphism in the IFN-γ gene, the latter being associated with severity of the disease.29 Linkage of specific TCR genes to inheritance of Hashimoto’s thyroiditis has also been reported. A specific TCR restriction fragment length polymorphism (RFLP) was increased in Hashimoto’s thyroiditis, as well as in Graves’ disease,30 and a TaqI RFLP for the Va gene of TCR was also increased.31 TCR Vbeta gene utilization was diminished, but selective expression was not observed in Hashimoto’s in contrast to Graves’ disease.32 Inheritance of specific allotypes of the immunoglobulin G (IgG) heavy chain is also seen in autoimmune thyroiditis.33 The CT60 polymorphism of CTLA-4 maps an important genetic determinant for the risk of Hashimoto’s disease across diverse populations34 and may identify patients with celiac disease who are at risk for Hashimoto’s disease and type 1 diabetes.35 Recently, X chromosome inactivation has been reported to be an important contributor to the increased risk that females may develop Hashimoto’s thyroiditis.36
Antibodies to Thyroid Antigens
THYROGLOBULIN
Experimental autoimmune thyroiditis with histologic findings similar to those of Hashimoto’s thyroiditis can be induced in animals by immunization with human thyroglobulin in an adjuvant. It can also be produced by depleting rats of T cells followed by thyroglobulin (Tg) administration. In both these animal models, strain specificity is vital, indicating the role of MHC class II-encoded susceptibility.37
Human Tg has at least 40 antigenic epitopes, but only one or two of these bind human TgAbs.38 Although the TgAb response in autoimmune thyroid disease (AITD) is typically polyclonal on isoelectric focusing,39 TgAb from patients with AITD are specific to human Tg and are directed toward a restricted number of epitopes on Tg, unlike rabbit TgAb, which recognizes Tg from other animal species.40 However, thyroglobulin is not isolated from the immune system in the thyroid follicles but is normally present in the circulation of humans.14 Thyroglobulin-binding lymphocytes can also be detected in the fetus.15
Evidence suggests that TgAb from patients with AITD recognize Tg from normal and AITD thyroids differently, suggesting the presence of antigenic variations between the Tg from healthy and AITD thyroids.41 TgAb in AITD is mainly IgG (which consists of four subclasses) rather than IgM, which is more likely to be present in healthy individuals.42 TgAb in AITD states is predominantly of the IgG4 subtype, and the weak complementing fixing property of TgAb is probably due to the predominance of this isotype, which is a poor activator of the complement cascade.43 This implies that antithyroglobulin antibodies are not directly related to tissue damage in Hashimoto’s thyroiditis. The pattern of Tg recognition, as assessed by the use of inhibition of Tg binding by four recombinant TgAb-Fab, showed that the pattern was similar when patients with Hashimoto’s disease were compared with patients with Graves’ disease.44
TgAb are found in patients with AITD but not consistently so, being present in only about 60% of patients.42 Unlike the thyroid peroxidase antibody (TPOAb), TgAb does not fix complement as stated above, probably because the epitopes on the large molecule are widely spaced and are unable to achieve the cross-linking necessary for complement activation. Further, TgAb is also found in healthy individuals with no evidence of thyroid disease.45 A role for TgAb in the pathogenesis of AITD therefore remains unproven.
THYROID PEROXIDASE
Thyroid peroxidase (TPO) evokes high-affinity, IgG-class autoantibodies (TPOAb) and TPO-specific T cells that are markers of thyroid infiltration or are implicated in thyroid destruction, respectively.46 Thyroid peroxidase (TPO), the major antigen in human Hashimoto’s disease, was shown to be identical to the previously termed microsomal antigen in the 1980s. TPOAbs in sera from patients with Hashimoto’s thyroiditis are predominantly polyclonal. Anti-TPO antibodies can induce complement-dependent cytotoxicity.47 In fact, antibodies against complement (anti-C1q) are found in patients with Hashimoto’s disease, and they correlate with thyroid-stimulating hormone (TSH) levels. Anti-C1q may be pathogenically involved in destruction in this disease independent of thyroid antibodies.48 Anti-idiotypic antibodies against TPO antibodies are occasionally found in the sera of patients with autoimmune thyroid disease and might be involved in the regulation of autoimmunity.49 A study of epitopic recognition patterns of TPOAb in healthy individuals and patients with Hashimoto’s thyroiditis has shown that specific immunodominant regions are associated with Hashimoto patients but not with normal controls. Whether the propensity to produce antibodies to certain TPO epitopes is of pathogenetic relevance is not clear.50 However, TPOAbs can damage thyroid cells as they activate the complement cascade. TPO itself appears to interact with TPO-specific T cells that are implicated in thyroid destruction. TPO antibodies that are transferred passively from mothers with Hashimoto’s thyroiditis do not seem to damage the thyroid or to affect thyroid function in the fetus or neonate.51
TPOAbs are positive in more than 90% of patients with Hashimoto’s thyroiditis, regardless of the presence of hypothyroidism or euthyroidism. The superior diagnostic value of TPOAb rather than TgAb for the confirmation of Hashimoto’s thyroiditis has led many hospitals and laboratories to rely solely on TPOAb measurements. This is satisfactory for more than 95% of patients, but in some instances, patients have only TgAb as a marker of thyroid autoimmunity. Currently, the clinical importance of anti-TPO antibodies lies in the diagnosis of thyroid autoimmunity, but T cell–mediated immunity to TPO is an ongoing field of investigation that should improve our understanding of the pathogenesis of Hashimoto’s thyroiditis.
THYROID-STIMULATING HORMONE RECEPTOR ANTIBODIES
Thyroid-stimulating hormone (TSH) stimulation-blocking antibody (TSBAb) can inhibit TSH action on thyroid and cause atrophic hypothyroidism in autoimmune thyroiditis.52 Although the specific epitopes for thyroid-stimulating antibody (TSAb) or TSBAb are still uncertain, the major epitope of TSBAb seems to be found in the C-terminal part of the extracellular domain (around 300 to 400 amino acids),53 probably in close proximity to that for TSAb.53 However, in vitro conversion from TSBAb to TSAb after the addition of antihuman IgG antibody suggests that TSAb and TSBAb are not determined solely by their epitopes, and the same TSHR antibody might act as a stimulator or a blocker, depending on the influence of other factors.54 TSBAb are uncommon as a cause of immune-mediated hypothyroidism, although they have been noted in a few patients with goitrous autoimmune thyroiditis, as well as in patients with atrophic chronic thyroiditis.52 Regional variations in prevalence have been noted, with the antibody reported more frequently in Japan.
OTHER ANTIBODIES
The Na+/I− symporter (NIS), a membrane glycoprotein, mediates iodine uptake into the thyroid follicular cell. Although reports have described antibodies to this moiety in Hashimoto’s thyroiditis, evidence that NIS is a major autoantigen now is not generally accepted. For example, anti-NIS antibodies were found to be positive in only 15% of patients with Hashimoto’s thyroiditis.55
Antibodies to thyroxine (T4) and triiodothyronine (T3) sometimes are found in patients with autoimmune or other thyroid diseases. They are seen in 14% and 35%, respectively, of patients with primary hypothyroidism, in most of whom TGAbs are found in high titer.56 The pathogenetic significance of these antibodies is not known. Probably, they are of little importance as long as the thyroid can produce enough thyroid hormone to sustain adequate serum levels of free hormone. These antibodies interfere with measurements of serum T4 and T3, especially in assays of free T4 and free T3.57 Antibovine TSH autoantibodies, occasionally found in Graves’ disease, are also reported in Hashimoto’s thyroiditis.58 Their pathogenetic significance is unclear. They are speculated to be anti-idiotypic antibodies to anti-TSH receptor antibodies (TRAbs) in Graves’ disease. They may interfere with the measurement of TRAbs and yield unusually high or negative titers. Autoantibodies against several other thyroid components have been reported. Antibodies to colloid antigen-2, distinct from thyroglobulin, have been detected. Antibodies to cell-surface antigen (distinct for TPO) are detected by the patchy immunofluorescent staining of the cell surface or by mixed hemabsorption.
Considerable interest has been expressed in past years in the concept of direct growth-stimulating antibodies or growth-blocking antibodies found in goitrous Hashimoto’s thyroiditis and in primary myxedema, respectively.37 However, contoversy regarding the assay has impeded progress.59 Following the development of a sensitive bioassay for growth-stimulating antibody, which did report positive findings in goitrous Hashimoto’s disease, no studies have explored the use of this assay in greater detail. Autoantibodies against other cellular components, so-called natural antibodies (not always thyroid cell-specific), have also been reported, for example, antibodies to tubulin and calmodulin60 and the ganglioside asialo-GM1, present in the plasma membrane of human thyroid.61 Measurement of natural antibodies (to DNA, actin, myoglobin, myosin, trinitrophenyl hapten, and tubulin) has shown that around 50% of a series of Hashimoto’s patients are positive for one or more of these antibodies, and this positivity appears to correlate with thyroid antibody activity.62 Antibodies to other organs related to autoimmune disease (e.g., islet cells, adrenal cortex, gastric mucosa, parathyroid) are found in autoimmune thyroiditis in higher incidence than in the general population.60 (See section on other autoimmune diseases.)
Cellular Abnormalities
The breakdown of immunologic self-tolerance leads to the presentation of self antigens and the expansion of autoreactive T cells. Consequently, release of inflammatory cytokines and differentiation of B cells producing antibodies occur. The T cells in Hashimoto’s disease are of the type 1 helper T cell (Th1) type, characterized by the production of IL-2 and IFN-γ.
It has become clear that regulatory T cells (T-reg) (with CD4+/CD25 surface expression) play an important role in the maintenance of immune tolerance and the prevention of its breakdown. T-reg express FoxP3, a specific gene marker for the inhibition of expression of inflammatory cytokines. Hence, in vivo depletion of this subset exacerbates autimmune disease in the Hashimoto’s mouse animal model. The development of transgenic animal models of autoimmune thyroiditis has suggested that T-regs have a role in the progression of Graves’ disease to Hashimoto’s thyroiditis and subsequent hypothyroidism.63 In addition to the predominance of the Th1 lymphocyte subset, the B7-1 (CD80) molecule preferentially acts as a co-stimulator for the generation of Th1 cells and has been recognized on thyrocytes in Hashimoto’s disease.64 This phenomenon, together with expression of MHC class II and intercellular adhesion molecule (ICAM)-1,65 leads to T cell differentiation from Th0 to Th1 cells and maintenance of thyroid autoimmunity. Thyroid cells in autoimmune thyroid disease also produce several cytokines (e.g., IL-1, IL-6, tumor necrosis factor-β [TNF-β]) that influence lymphocytic responses (vide infra).
In studies of intrathyroid-infiltrating lymphocytic populations, T lymphocytes predominate over B cells and CD8+ T lymphocytes are increased in Hashimoto’s thyroiditis. Analysis of the gene for the variable region of the a chain (Va gene) of the TCR suggested that the infiltrating T cells are a highly restricted population,66 raising the possibility of a clonal restriction in this autoimmune disease. However, this has not been confirmed.67 Several in vitro cytotoxic mechanisms have been demonstrated. Cytotoxic T cells that are specific for thyroid epithelial cells are observed and antibody-mediated cell cytotoxicity activity can be found in the sera of patients with Hashimoto’s disease. In addition, complement-dependent cytotoxic antibodies are found in sera from these patients, and natural killer lymphocyte counts correlate inversely with thyroid hormone levels. These factors undoubtedly contribute to thyroid follicular cellular damage. Their importance in the whole destructive cascade has been overshadowed to some extent by the data related to apoptosis of thyroid cells (vide infra).
Cytokines
Cytokines are known to have a wide variety of inflammatory and immunomodulatory effects; therefore, it might be thought that many steps in thyroid autoimmunity are mediated and/or modulated by cytokines.68
Characterization of T cells into Th1 and Th2 based on different patterns of cytokine production has aided the understanding of immunologic events in Hashimoto’s disease, although the picture in the human is not as clear as in animals. Cytokines associated with Th1 and Th2 are shown in Table 83-4. In general, the pattern of T cell response in autoimmune hypothyroidism is a Th1 pattern. Following activation by autoantigen recognition, thyroid-specific lymphocytes produce IL-2 with induction of T cell proliferation and differentiation. IFN-γ is also produced and natural killer cells activated. Thyroid autoimmunity occurs in humans who received IL-2 or IFN-γ for the treatment of cancer or viral hepatitis.69,70 A broader range of cytokines then is produced from these activated lymphocytes, resulting in amplification and perpetuation of the immune response. In addition, some cytokines stimulate human lymphocyte antigen (HLA) class II expression on thyroid epithelial cells, which may be important for the amplification and progression of the disease. Data from in vitro effects of cytokines on cultured thyroid cells also indicate that some cytokines (e.g., TNF-α, which is mainly produced by monocytes) may be directly cytotoxic to thyroid cells in vivo. The mechanisms of cytokine interaction are not simple. For example, autoantibodies and complement may promote mixed Th1/Th2 cell cytokine response by enhancing the uptake of autoantigens by antigen presenting cells.71 Another molecule, CTLA-4, whose gene is now a known risk factor for autoimmune thyroid disease, is expressed by activated T and B lymphocytes. Levels of serum CTLA-4 are elevated in autoimmune thyroid disease, and this may play a pathogenetic role in this condition.72 It should be noted that some cytokines (e.g., IL-1, IL-6) are produced by thyrocytes, as well as by lymphocytes. Thus, interleukins produced by thyrocytes would stimulate intrathyroidal lymphocytes. IL-1 stimulates T cells to release lymphokines and has many other inflammatory effects. IL-1 can induce thyrocyte apoptosis through Fas-Fas ligand interaction (vide infra).
Table 83-4. Cytokine Pattern from Th1 and Th2 CD4+ Cells
| Th1 | Th2 | |
|---|---|---|
| IL-1 | ++ | ++ |
| IL-2 | +++ | − |
| IL-4 | − | +++ |
| Il-5 | − | +++ |
| Il-10 | + | +++ |
| IL-13 | − | +++ |
| IFN-γ | +++ |
IFN, Interferon; IL, interleukin.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


