PATHOGENESIS
From a histologic point of view, two types of nodules (Figs. 82-1 and 82-2) may be discerned: a monoclonal and a polyclonal type. Studer and his group developed the concept that even if monoclonal at the molecular level, nodules may become polyclonal from a functional and histologic aspect during evolution. They suggest that individual follicular cells may acquire new qualities that were not present in the mother cells but become inheritable during further replication. Obviously this change supposes some sort of genetic event. This sequence of events may lead to loss of anatomic and functional integrity of the follicular cells. The process might be accelerated by stimulatory factors such as TSH (e.g., in iodine deficiency, by goitrogens) and by local stimulatory and growth factors.6 (See also Chapter 87).
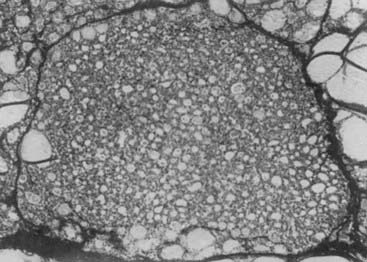
FIGURE 82-1. Uniform nature of cells formed in a nodule by proliferation of only one or a few clones of epithelial cells.
(From Studer H, Ramelli F: Simple goiter and its variants: euthyroid and hyperthyroid multinodular goiters. Endocr Rev 3:40,
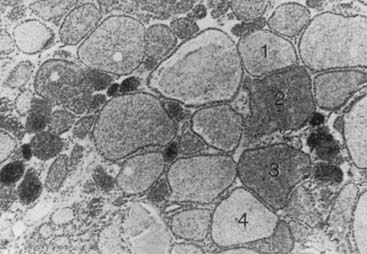
FIGURE 82-2. Autoradiograph of a hot nodule, illustrating areas with different capacity of uptake of radioiodine.
(From Studer H, Gerber H, Peter HJ: Multinodular goiter. In DeGroot LJ [ed]: Endocrinology, 2nd ed, vol 1. Philadelphia: WB Saunders, 1989, p 722.)
At the genetic level, two types of monoclonal autonomously functioning nodules have been reported. Both are somatic mutations. One involves the TSH receptor (TSHR) gene and the other involves the Gsα protein gene. Both mutations lead to constitutive activation of the adenylate cyclase system, probably also activation of the inositol phosphates pathways7 in the follicular cell, and to autonomy of the follicle. Mutations of the TSHR gene, and with a much lower prevalence, the Gsα gene, play a major (principal) role in the pathogenesis of AFTN.8 For those monoclonal toxic adenomas (TA) in which no mutations are found in the TSHR or Gsα unit, probably other somatic mutations are involved.9 In a recent study of 75 hot thyroid nodules, somatic TSH receptor mutations were detected in 57% and Gsα mutations in 3% of the nodules.10 The same group studied females with AFTN, but without mutations in the TSH-R or Gsα unit, and detected a monoclonal origin in 10 out of 20 of cases when tested for X-chromosome inactivation. This strongly suggests a mutation at other location(s).10 Although some believe that iodine deficiency can increase mutation rate and functional expression of autonomy11 in AFTN, others12 consider the fundamental process of goitrogenesis in (multi)nodular goiter as independent from iodine deficiency but operating through mechanisms that are innate to the hereditary and acquired heterogeneity among the thyrocytes themselves. However, superimposed iodine deficiency may shift clinical expression to younger ages. Van Sande and co-workers13 hypothesized that as 16 different activating mutations were identified in the TSHR gene, this receptor is in a constrained conformation in its wild-type form. They also report that AFTA have a high level of Na+/iodide symporter gene expression, a high thyroperoxidase mRNA and protein content, and a low H2O2 generation. Inositol uptake was also increased, but inositol phosphates were not increased. TA secreted more thyroid hormone than the quiescent surrounding tissue. Other characteristics of TA were increased cycling of thyrocytes as compared to normal surrounding tissue, little apoptosis, and low expression of early immediate genes.14 An important study by Fuhrer and co-workers15 showed that a panel of different activating TSHR mutations caused different functional and morphologic responses in vitro in rat and human primary thyrocytes. Their data suggest that different biologic properties of the TSHR mutants may result in different in vivo phenotypes. Finally, other mutated genes causing AFTN may be located in the AMP cascade.
PATHOLOGY
On macroscopic examination, a solitary toxic nodule is surrounded by normal thyroid tissue that is functionally suppressed. Rarely, a microscopically monotonous picture is seen that consists of uniform follicular cells without signs of malignancy. Usually the picture is heterogeneous with cells of different size, sometimes with signs of fresh and old hemorrhage and calcification. This picture, however, does not exclude a monoclonal origin of the nodule but may be the result of stimulatory local growth factors (see the section entitled “Pathogenesis”). No information is available at present on the exact ratio between primarily monoclonal and polyclonal AFTNs of the thyroid. Sometimes, autonomously functioning micronodules are present in the surrounding thyroid tissue. This finding is in agreement with the thesis of Studer and co-workers6 that the true adenoma is one end of a large spectrum of thyroid nodules growing from single thyrocytes or tiny cell families, each replicating with an individual growth rate, whereas the grossly abnormal multinodular goiter is at the other end of the scale.
CLINICAL FEATURES
The signs and symptoms of patients with toxic adenoma are those of thyrotoxicosis, without the specific signs of Graves’ disease, such as eye signs, pretibial myxedema, and acropachy (see Chapter 80). On palpation of the thyroid region, a nodule is found in one of the lobes, usually with a diameter of 3 cm or larger.2 The thyroid tissue surrounding the nodule and the thyroid lobe on the other side are not usually palpable because of TSH suppression.
LABORATORY DIAGNOSIS
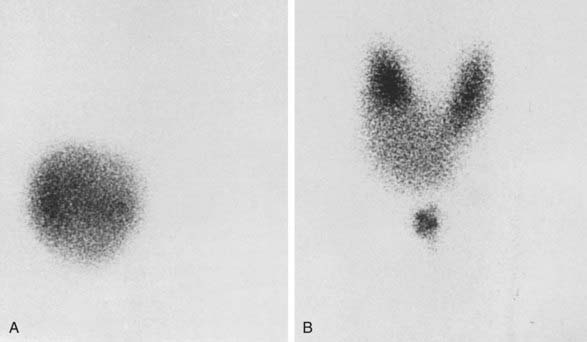
When a single nodule is found in the thyroid and the patient is clinically thyrotoxic, estimation of serum TSH is sufficient for the diagnosis of toxic adenoma. To have an idea about the severity of the thyrotoxicosis, estimation of serum free thyroxine (T4) is sufficient. If this parameter is normal, serum triiodothyronine (T3) or free T3 should be determined because it may be solely elevated in minimal thyrotoxicosis, that is, T3 toxicosis. If findings on palpation are inconclusive, it is wise to perform a scintiscan of the thyroid. In the case of a toxic nodule, activity is seen in the nodule, with minimal or no activity anywhere else in the thyroid region (Fig. 82-3). The presence of a thyroid carcinoma in an AFTN is rare.16 Although some authors are of the opinion that the occurrence of carcinoma in an AFTN may be more than coincidental,17 most thyroidologists believe that such is not the case. The differential diagnosis of a toxic nodule includes relapse of hyperthyroid Graves’ disease in remnant thyroid tissue after thyroid surgery or in thyroid dysgenesis. The latter possibility is especially remote. Theoretically, a TSH stimulation test would distinguish between an AFTN and the two other possibilities, but this test is hardly ever necessary in clinical practice. It is of no value to perform fine-needle aspiration cytology of an AFTN, because differentiation between a follicular adenoma and carcinoma is difficult if not impossible with this technique. When a hot nodule is present, that is, prominent uptake in the nodule but less in the surrounding tissue while serum TSH is normal, autonomous function can be tested by performing a T3 or T4 suppression test. In the case of autonomous function, uptake in the nodule is still present after the administration of 25 µg T3 three times daily for 10 days or 125 µg T4 for 14 days, whereas uptake is suppressed in the surrounding tissue. Sometimes uptake in a single nodule is indistinguishable from uptake in the surrounding tissue, a situation described as a “warm” nodule. This picture may be generated either by normal affinity of the nodular tissue for the isotope or because a nonfunctioning nodule is surrounded by normal thyroid tissue. Thus the possibility of the presence of a carcinoma in a warm nodule is certainly not excluded, which means that it should be evaluated as though it were a cold nodule (see Chapter 89).
TREATMENT
No treatment of a hot nodule is necessary as long as the patient remains euthyroid. Regular TSH measurement at intervals of a half to 1 year suffices. Most patients remain euthyroid. Occasionally, a hemorrhage in the nodule leads to spontaneous resolution. Treatment of toxic nodules with antithyroid drugs is useless because after discontinuation of medication, relapse invariably occurs. Three modes of treatment of toxic nodules include nodulectomy, administration of radioactive iodine, and percutaneous injection of alcohol into the nodule. Nodulectomy is very effective in rendering the patient euthyroid and has a low surgical complication rate. Surprisingly, permanent hypothyroidism develops in about 5% of patients, perhaps because of coexistent thyroid disease.2,18 Obviously, the disadvantages of surgery are its operative risks, the residual scar, and its cost, which is high in relation to the other two forms of treatment.
Treatment with radioactive iodine is safe, cheap, and effective. Two reports indicate no risk of posttreatment hypothyroidism 6 months after treatment in a total of 93 patients.19,20 However, when a longer period of follow-up was taken into account in 23 patients, such as 4 to 16.5 years after treatment, hypothyroidism developed in up to 36%.21 In another study of 126 patients with autonomously functioning nodules, the percentage of hypothyroidism after a mean period of 10 years was 9.7% when the nodules were hot and 1.5% when toxic.22 In this and the previous study, no relationship was found between the total dose administered, the size of the nodule, and the development of hypothyroidism. However, if thyroid autoantibodies were present, hypothyroidism occurred in 18% of patients and, when absent, in 1.4%.22 It is important that when 131I is administered, uptake be present only in the nodule and not in the surrounding tissue or in the other lobe, which could occur after pretreatment with antithyroid drugs. To avoid this complication, pretreatment with T3 or T4 might be considered. In some patients, typical Graves’ disease has developed months after treatment of a toxic nodule with 131I. Possibly, antigens released by the 131I therapy induce or exacerbate an autoimmune response.
The third treatment modality is of rather recent date and involves percutaneous alcohol injection into the nodule. Euthyroidism is achieved in 65% to 85% of patients by 12 months after treatment. Injections are repeated 2 to 12 times at weekly intervals. The treatment is usually well tolerated, with few side effects. When nodule volume exceeds 30 mL, results are less favorable.23,24 All three treatment modalities are acceptable, the choice depending on local circumstances and the patient’s preference. Laser photocoagulation of AFTN is the most recent introduced therapy.25 A recent study showed that its efficacy is comparable to that of ethanol injection, with possibly less ensuing hypothyroidism.24
Silent or Painless Thyroiditis
Silent thyroiditis is an autoimmune thyroiditis that usually comes to clinical attention because of symptoms of thyrotoxicosis caused by leakage of thyroid hormone from a painless thyroid gland. The condition often occurs in the postpartum period.
INCIDENCE
The incidence varies geographically. In 1980, this syndrome accounted for 10% of cases of thyrotoxicosis in Japan but only 3% to 4% in New York City.22,26,27 A random poll showed that silent thyroiditis was uncommon in Argentina, Europe, and the east and west coasts of the United States but occurred more frequently around the Great Lakes and in Canada.28 Patients are usually between 30 and 60 years old, and the female-to-male ratio is 1.5 to 1. Apart from its association with pregnancy, known then as postpartum thyroiditis, the condition is currently rarely diagnosed. Postpartum thyroiditis occurs in 5% to 9% of women in the first year after delivery, especially in those who have circulating autoantibodies against thyroperoxidase.29
ETIOLOGY
Silent thyroiditis is an autoimmune lymphocytic thyroiditis, and many patients with this disease have a family history of thyroid disease. Postpartum thyroiditis has been significantly associated with HLA-D3 and HLA-D5.30 Exposure to iodine, such as in the form of amiodarone or lithium, interleukin (IL)-2, interferon, and etanercept has been suggested as an initiating event.31–33 Silent thyroiditis is associated with other autoimmune diseases such as rheumatoid arthritis, systemic sclerosis, Graves’ disease, primary adrenal insufficiency, systemic lupus erythematosus, idiopathic thrombocytopenic purpura, rubella, and seasonal allergies.33–38
PATHOLOGY
Microscopically, follicles are disrupted, and infiltration of lymphocytes and plasma cells occurs. The infiltration may be focal or diffuse, and sometimes formation of lymphoid follicles is seen. Follicular cells may be cuboidal or, when stimulated by TSH in the hypothyroid phase, columnar. Sometimes Hürthle or Askanazy cells are present. These cells are large and oxyphilic and contain many mitochondria. Thyroid tissue obtained during hypothyroidism or the recovery phase shows regenerating follicles with little colloid. Occasionally, persistent lymphocytic infiltration is observed. Extensive fibrosis may ultimately develop. A few multinucleated giant cells are regularly present.39
CLINICAL FEATURES
The initial symptoms in 112 patients with 122 episodes of silent thyroiditis have been reviewed.40 Characteristically, thyroid pain was absent in all cases. The female-to-male ratio was 1.3 : 1. The mean age (±SD) in females and males was 32 ± 8.5 and 24.9 ± 8.2 years, respectively. Recurrences were uncommon. The symptoms are similar to other causes of thyrotoxicosis and varied from mild to severe. Specific signs that are characteristic of Graves’ disease, such as eye signs, pretibial myxedema, and acropachy, were absent. The mean duration of the toxic phase in these patients was 3.6 ± 2.0 months. In most reports, the thyroid gland had a firm consistency. In about half the patients, a goiter was present. The course of the disease follows four sequential stages: thyrotoxicosis, euthyroidism, hypothyroidism, and euthyroidism. These stages need not be present in all patients. In one series, 57 of 112 patients became euthyroid, and hypothyroidism did not develop. Clinical hypothyroidism was present in only 32 patients. Hypothyroidism was transient in 24 patients but permanent in 8, who required thyroid hormone substitution. Ultimately, about half of the patients with silent thyroiditis become permanently hypothyroid.41
LABORATORY FINDINGS
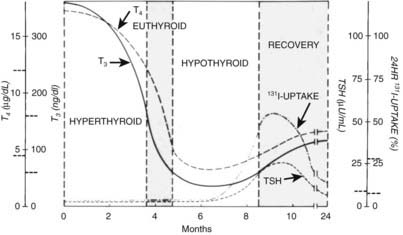
The acute (first) phase of the disease is characterized by leakage of thyroid hormone and thyroglobulin from the damaged thyroid. This leakage results in elevated serum concentrations of thyroid hormones and thyroglobulin and suppression of serum TSH. Uptake of radioactive iodine by the thyroid is absent in this stage. The C-reactive protein (CRP) and the erythrocyte sedimentation rate (ESR) are mostly, but not always, slightly elevated. The ESR was elevated in 34 of 53 episodes but higher than 40 mm only in 8.40 This result contrasts with subacute thyroiditis, in which the ESR is invariably much more elevated. T4 and T3 start to decline in the first phase and reach normal levels in the second (euthyroid) phase, but TSH remains suppressed. In the third (hypothyroid) phase, thyroid hormone levels are subnormal, and serum TSH starts to rise at the end of this stage. Because of TSH stimulation, the fourth stage is characterized by normalization of serum T4 and T3. Serum TSH ultimately normalizes, but normalization can take several months, so temporary subclinical hypothyroidism intervenes. For the mean period of the different phases, see Fig. 82-4.
TREATMENT
The degree of thyrotoxicosis is usually mild in silent thyroiditis, and treatment is not usually necessary. Prescription of α-adrenergic blocking agents may be considered. Antithyroid drugs have a very limited role because the thyrotoxicosis is not the result of increased thyroid hormone synthesis. Propylthiouracil or ipodate to block peripheral conversion of T4 to T3 may be of some value. In more serious cases, prednisone in a dose between 30 and 60 mg/day has a rapid ameliorating effect. The dose should be continued for 1 to 2 weeks and then be slowly tapered.42 In case of relapsing thyroiditis, prednisone can be reinstituted. It is seldom necessary to perform thyroidectomy. If necessary, radiochemical “thyroidectomy” may be contemplated during remission when sufficient thyroid uptake of radioactive iodine is present for effective treatment, often in the presence of prednisone administration. As was stated, thyrotoxicosis is usually mild, and “definitive” treatment is seldom necessary. After the thyrotoxic phase, temporary hypothyroidism develops in about 40% of patients. If needed, thyroid hormone treatment can be instituted in a dose that allows TSH to remain mildly elevated to promote resumption of thyroid hormone synthesis in the recovery phase. Only a small proportion of patients (see previous discussion) need permanent and full-dose substitution at this stage. Finally, however, permanent hypothyroidism develops in about half of the patients. This result is in contrast to subacute thyroiditis, after which patients almost always become permanently euthyroid. Thus patients who suffered from silent thyroiditis need lifelong follow-up because hypothyroidism may develop years later.43
Thyrotoxicosis Factitia
Thyrotoxicosis factitia is primarily a psychiatric disorder. Patients surreptitiously ingest thyroid hormone in excessive amounts. When confronted with the situation, they usually deny doing so. Physicians should be aware of the phenomenon, or the diagnosis might be missed. Patients are usually overtly thyrotoxic but do not show eye signs, except those of sympathetic overactivity, such as eyelid retraction.
Other signs of Graves’ disease such as eye signs, pretibial edema, acropachy, and goiter are also absent. Differentiation from Graves’ disease is also feasible by color Doppler sonography, which shows absent thyroid vascularity and low-normal peak velocity, whereas these signs are increased in Graves’ disease.44 Because of TSH suppression, the thyroid shrinks and is often not palpable. Thyroid uptake of radioactive iodine is absent. Serum thyroglobulin is low or below detection limits in thyrotoxicosis factitia but elevated in silent thyroiditis. Factitious thyrotoxicosis is not difficult to distinguish from toxic multinodular goiter or toxic adenoma. Differentiation from subacute thyroiditis is easy on clinical grounds, because these patients suffer from frequent severe pain in the thyroid region. Furthermore, the CRP or ESR and serum thyroglobulin concentration are elevated in subacute thyroiditis. The diagnosis of thyrotoxicosis factitia should be considered when laboratory results are contradictory. Psychiatric help is urgently needed for such patients.
Thyrotoxicosis caused by accidental intake of excessive amounts of thyroid hormone has been observed in the “hamburger toxicosis patients.” Two epidemics were caused by the inclusion of bovine thyroid in hamburger.45,46
Thyrotoxicosis Caused by Pregnancy and Trophoblastic Disease
Human chorionic gonadotropin (hCG) has intrinsic TSH-like activity. In about 2% to 3% of normal pregnancies, gestational transient thyrotoxicosis (GTT) is present because of elevated hCG serum concentrations. Familial gestational hyperthyroidism has recently been described and is caused by a missense mutation in the TSHR that renders it hypersensitive to hCG. Thyrotoxicosis may also be induced by molar pregnancy and by trophoblastic disease in men and women.
TSH-LIKE ACTIVITY OF HUMAN CHORIONIC GONADOTROPIN
The hCG from concentrated human pregnant urine has weak TSH-like activity when tested in a mouse bioassay.47 The hCG that is purified from molar tissue has intrinsic TSH bioactivity in the same bioassay, though 4000 times less than that of human TSH on a molar basis.48 However, when produced in sufficient amounts, it may induce clinical hyperthyroidism in humans, as shown in 2 of 20 patients with gestational trophoblastic neoplasia.49 These patients had extremely high serum (3,220,000 and 6,720,000 IU/L) and urine concentrations of hCG that correlated closely with TSH-like bioactivity. Other patients with moderately elevated serum hCG levels, between 110,000 and 310,000 IU/L, were euthyroid. When tested on human thyroid cell membranes, 1.0 IU hCG is biologically roughly equivalent to 0.27 µIU hTSH.50 Both hCG and human luteinizing hormone (HLH) compete with TSH for the TSHR, and hLH also has weak (10 to 100 times higher than hCG) TSH-like activity.50–52 The β subunit of hCG and hLH share 85% sequence identity in the first 114 amino acids but differ in the carboxyl-terminal peptide because hCG-β contains an extension of 31 amino acids.53 Carboxypeptidase digestion of hCG, with amino acid residues 142 to 145 cleaved from the β subunit, leads to an increase in its capacity to stimulate adenylate cyclase in human thyroid membranes.54 A variant of hCG that is lacking the C terminus of the β subunit because of enzymatic cleavage has been identified in pregnancy serum and molar tissue.55 In studies using human thyroid membranes56 or a cell line transfected with human TSHR,57 desialylated forms of hCG exhibited stronger inhibition of TSH-mediated cyclic adenosine monophosphate responses than did native hCG. Both TSH binding and TSH-induced adenylate cyclase stimulation were found to be more effectively inhibited by desialylated variants of hCG than unmodified hCG was.58 From these and other studies it seems that the biologic effect of hCG is predominantly confined to hCG containing little or no sialic acid. In cultured FRTL-5 cells, hCG has been found to increase iodide uptake, and it also causes a dose-related increment in adenylate cyclase activity and thymidine uptake.59,60
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


