Subclinical autoimmune thyroiditis may become clinically apparent in the postpartum period as the result of an exacerbation of the autoimmune process for reasons that are not yet clear. Typically, such women have TPO autoantibodies antepartum and experience transient thyroid dysfunction (thyrotoxicosis followed by hypothyroidism, or either phase alone) accompanied by a small painless goiter, with full recovery within a year after delivery.6 It is now known that postpartum thyroiditis is a risk factor for future permanent hypothyroidism, which ensues in 20% to 30% of cases in the subsequent 5 years. Around 5% of pregnant white women have one or more episodes of postpartum thyroiditis.
Overt hypothyroidism caused by autoimmunity has two main forms: Hashimoto’s (or goitrous) thyroiditis and primary myxedema, also known as atrophic thyroiditis. The former is characterized by a variably sized, firm goiter, often with an irregular surface; the goiter is typically painless, although a rare, painful variant occurs that, in contrast to subacute thyroiditis, might respond poorly to steroid treatment.7 Such patients may have a goiter and normal thyroid function or subclinical or overt hypothyroidism, depending on the extent of the autoimmune destructive process. However, in primary myxedema, the typical manifestation is clinically evident hypothyroidism, obviously without a goiter. Many attempts have been made to ascribe distinct genetic or pathogenic factors to these two disorders, but in most cases, a unique pathogenesis is not evident, and it seems most likely that they are simply at opposite ends of a spectrum of clinical features, with gradual diminution in the size of the goiter noted as disease progresses.
Similarly, Graves’ disease shares many immunologic features with autoimmune hypothyroidism, and even the hallmark of Graves’ disease, TSHR autoantibodies, can be found in some patients with autoimmune hypothyroidism, although their effects are masked by a more vigorous destructive autoimmune process (see below). Graves’ disease is the most common autoimmune disorder in the United States, with an estimated weighted prevalence of 1.2 per 100, 88% of whom are women.5 Many of these individuals progress to hypothyroidism, either spontaneously after successful treatment with antithyroid drugs or iatrogenically after radioiodine therapy or surgery.
Additional clinical details on these conditions are given in subsequent chapters; this chapter provides an overview of the basic causative and pathologic factors involved in autoimmune thyroid disease.
Pathology
The typical appearance of Hashimoto’s thyroiditis is termed struma lymphomatosa to indicate the extensive infiltration of the thyroid by lymphocytes, plasma cells, and macrophages.7 As well as a diffuse infiltrate, germinal center formation may be particularly prominent, and giant (Langerhans) cells can occur. The thyroid follicular cells are destroyed to a variable extent, depending on the chronicity of the disease, and during this process, the remaining cells become hyperplastic and undergo oxyphil metaplasia, which gives rise to the so-called Askanazy or Hürthle cells. Variable fibrosis and, in rare cases, concurrent changes typical of Graves’ disease appear—so-called hashitoxicosis.
Primary myxedema is characterized by extensive fibrosis, loss of normal lobular architecture, and gland atrophy; lymphocytic infiltration varies from minor to moderate. It remains to be established how frequently Hashimoto’s thyroiditis progresses to primary myxedema. The extent of fibrosis in autoimmune hypothyroidism is directly correlated with the age of the patient,8 compatible with the occurrence of such a progression, although little change in the pathologic appearance has been found in sequential biopsies over 10 to 20 years in those with Hashimoto’s thyroiditis.9 The histologic appearances of postpartum thyroiditis and silent thyroiditis resemble Hashimoto’s thyroiditis, although oxyphil metaplasia and germinal centers are less frequent, whereas in focal thyroiditis, mild Hashimoto-like changes are seen in localized areas of the thyroid, but most follicles are preserved.
The pathologic features of Graves’ disease usually are obscured by prior treatment with antithyroid drugs, whose effects on the autoimmune process are considered later. Hypertrophy and hyperplasia of the thyroid follicles may be noted, the epithelium is columnar, and the colloid shrinks.7 In addition, a variable degree of lymphocytic infiltration is present, sometimes with germinal center formation. Lymphoid hyperplasia also can be found in the thymus, lymph nodes, and spleen. Follicular involution and reversal of the lymphocytic infiltrate and hyperplasia occur with antithyroid drug treatment.
Thus, all forms of thyroid autoimmunity are associated with a lymphocytic infiltrate in the thyroid, and these lymphocytes are largely responsible for generating both T and B cell–mediated autoreactivity, although other sites such as thyroid-draining lymph nodes and bone marrow contain thyroid-autoreactive lymphocytes in autoimmune thyroid disease.10 Many investigations have used peripheral blood lymphocytes from patients, but although these cells are readily accessible, they reflect the behavior of only a small proportion of autoreactive lymphocytes migrating in the blood compartment in what is essentially a localized disease.
The Basis of Autoimmune Disease
This section provides only an attenuated overview of the basic mechanisms underlying autoimmunity; the reader is referred elsewhere for a more comprehensive review.11 For the purposes of this chapter, however, a brief summary of the normal immune response is followed by a description of how autoreactivity can arise.
THE NORMAL IMMUNE RESPONSE
The essence of the immune response is shown in Fig. 79-1. The initial step involves presentation of an antigen by an antigen-presenting cell (APC), such as a dendritic cell or macrophage, to a helper T cell, which recognizes the antigen by means of a specific T cell receptor (TCR).12 Such T cells can be identified phenotypically by expression of the molecule CD4 (CD stands for cluster of differentiation and is used to define an array of surface molecules on lymphocytes and other cells). The antigen is first taken up by simple phagocytosis or by involvement of specific surface receptors, and it is processed by the APC so that fragments of 12 to 18 amino acids are generated. These fragments are the “epitopes” of the antigen, which binds to major histocompatibility complex (MHC) class II molecules expressed constitutively by the APC. The MHC (termed HLA in humans and H-2 in mice) is a huge complex of genes that is of fundamental immunologic importance because class I (described below) and class II genes encode polymorphic molecules capable of binding a wide range of antigenic peptides.13 The ability of an individual to mount an immune response to any given antigen depends in part on the inheritance of class I and II genes, which determine (1) whether appropriate T cells develop in the thymus, because MHC molecules can positively or negatively select T cells with particular antigenic specificities, as determined by their TCR and stage of development; and (2) whether an antigenic epitope can bind to an appropriate MHC molecule and therefore be presented to a mature T cell in adult life.
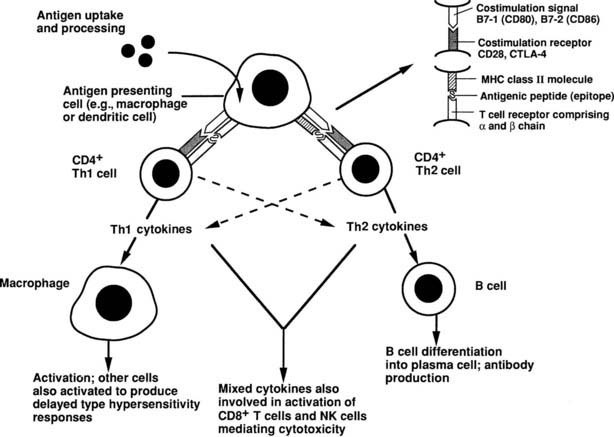
FIGURE 79-1. Simplified diagram of the key elements in a normal immune response, starting with antigen presentation. The type of immune response that is induced depends on the cytokine profile of the T helper cell that is stimulated.
(From Weetman AP: Recent progress in thyroid autoimmunity: an overview for the clinician. Thyroid Today 19[2]:1–9, 1996.)
Formation of the trimolecular complex between the MHC class II molecule, antigenic epitope, and TCR is followed by activation of the CD4+ T cell, a process that involves expression of the interleukin-2 (IL-2) receptor and autocrine stimulation by IL-2 release and leads to T cell proliferation, secretion of other cytokines, and thus the development of effector function. However, many other molecules are involved in this interaction and act as stabilizers, signal transducers, or providers of so-called costimulatory or second signals (Fig. 79-2). In brief, interaction of a number of adhesion molecule ligands and receptors, such as intercellular adhesion molecule-1 (ICAM-1, CD54)/lymphocyte function-associated antigen-1 (LFA-1, CD11a/CD18) and LFA-3 (CD58)/CD2, allows initial binding of the T cell to an APC. CD4 interacts with a nonpolymorphic region on the MHC molecule to stabilize the trimolecular complex; this activity explains why helper T cells recognize antigen that is presented by class II rather than class I molecules. CD3 is composed of five peptides that are expressed uniquely by all T cells and serves to initiate the intercellular events after TCR ligation. Finally, a host of costimulatory signals can determine whether antigen recognition proceeds or is terminated because of the absence of an essential costimulator or the presence of an inhibitory signal.
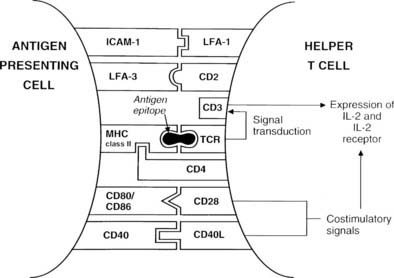
FIGURE 79-2. Diagram of the key molecular interactions in CD4+ T cell activation by an antigen-presenting cell (APC).
The best defined costimulatory pathway involves the interaction of B7-1 (CD80) and B7-2 (CD86) on the APC with CD28, which transduces a stimulatory second signal, or cytotoxic T lymphocyte antigen type 4 (CTLA-4, now named CD152), which transduces an inhibitory second signal on the surface of the T cell.14 Interaction of the TCR on naïve T cells with an MHC class II molecule plus epitope, in the absence of CD28 ligation, induces anergy (rather than stimulation) in the T cell, that is, the T cell is paralyzed and is unable to respond (Fig. 79-3). Similarly, engagement of CTLA-4 results in T-cell anergy. As is discussed below, induction of anergy is an important mechanism in preventing autoimmune responses. Other important membrane-bound costimulatory signals exist, for instance, ICOS (inducible costimulator) ligand, which seems to be particularly important for eliciting responses from previously activated (or memory) T cells.15 Thus, strikingly different outcomes are possible after antigen presentation, depending on which costimulatory signals are delivered, and the signal that is delivered in turn depends on the maturity of the T cell and the type of APC involved.
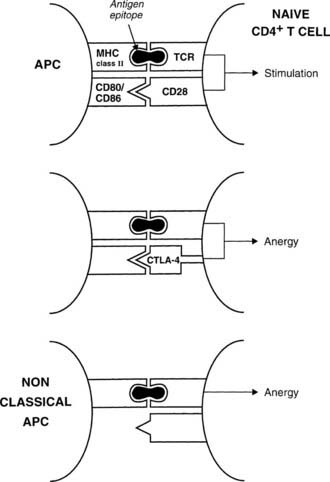
FIGURE 79-3. Mechanisms for anergy induction. The normal pathway for antigen presentation is shown in the upper panel; failure to provide an appropriate costimulatory signal (lower panels) results in anergy. Classic antigen-presenting cells (APCs) express costimulatory molecules, but class II–positive nonclassic APCs (e.g., thyroid cells) do not.
Activated CD4+ T cells can follow two broad pathways of function depending on their pattern of cytokine secretion (Table 79-2). Type 1 helper T (Th1) cells mediate delayed-type hypersensitivity responses, essentially the kind of destructive process that is seen in organ-specific endocrinopathies, whereas Th2 cells promote antibody production.13 A number of factors, including TCR affinity and ligand density, the nature of the APC, and the presence of non–T-cell–derived IL-4 and IL-12, determine which pathway is followed.16,17 Although this paradigm is undoubtedly useful, many helper T cells, particularly in humans, cannot be neatly classified according to the Th1/Th2 dichotomy. For example, a third subset, Th0 cells, produces a mix of Th1 and Th2 cytokines and is thought to be a precursor population; evidence suggests linear development of Th1 into Th2 cells via Th0 in humans.18 Another recently identified subset, Th17, produces the cytokine IL-17 and is highly proinflammatory, with a potent role in causing autoimmunity.17 Expanded clones of T cells arising from naïve T cells after ligation of the T cell receptor give rise to memory T cells, which help antibody production, promote inflammation, or regulate immune responses.19
Table 79-2. Characteristics of Murine CD4+ T Helper (Th) Cell Subsets; Similar but Not Identical Profiles Have Been Identified in Humans
| Characteristic | Th1 | Th2 |
|---|---|---|
| Cytokine Profile | ||
| IL-2 | ++ | − |
| IL-3 | ++ | ++ |
| IL-4 | − | ++ |
| IL-5 | − | ++ |
| IL-6 | − | ++ |
| IL-10 | − | ++ |
| Interferon-γ | ++ | − |
| Tumor necrosis factor | ++ | + |
| Lymphotoxin | ++ | + |
| Function | ||
| Delayed-type hypersensitivity | ++ | − |
| B-cell help | + | ++ |
| Eosinophil/mast cell production | − | ++ |
IL, Interleukin.
CD8+ T cells are typically cytotoxic but have in the past been assigned suppressor functions. Th1 and Th2 responses are mutually inhibitory; therefore, many suppressor phenomena may be due to a population of cells that are capable of secreting appropriate cytokines that can switch off an ongoing immune response (termed immune deviation). The best defined regulatory T cell subset is CD25+ CD4+, Foxp3+ and is generated in the thymus; the emergence of autoimmune disease in animals when such thymic development is interfered with provides compelling evidence for the importance of these cells.20,21 The cytotoxic function of activated CD8+ T cells is directed against antigenic epitopes that are synthesized within the target cell and presented by MHC class I molecules; the two classic groups of antigens that are recognized by these cells are the products of viral infection or malignant transformation.13 Destruction of target cells is mediated by two mechanisms: (1) the granule exocytosis pathway, which utilizes perforin and granzyme A and B; and (2) apoptosis, or programmed cell death. In the latter, engagement of Fas on the surface of the target cell with Fas ligand (FasL) on the T cell leads to activation of a chain of intracellular events that results in target cell apoptosis (Fig. 79-4). Fas expression is generalized, whereas FasL is restricted to cells of the immune system and to sites of immune privilege, as is described in the next section.
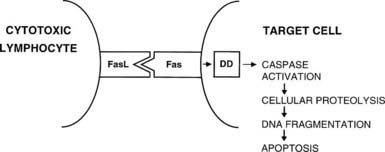
FIGURE 79-4. Interaction between Fas ligand (FasL) on cytotoxic lymphocytes and Fas on a target cell leads to signaling via the death domain (DD) proteins and apoptosis.
Mention should also be made of a further subdivision of T cells. Most T cells have a TCR that is a heterodimer composed of an α and a β chain. The genes for these chains are rearranged to ensure adequate diversity for antigen recognition. However, a small proportion of T cells have a TCR composed of a γ and a δ chain, and these receptors have a more restricted diversity.22 The role of γδ T cells is unclear, but they may be important in mucosal immunity and protection against particular microorganisms.
Two other cell types are essential components of the immune response. B cells produce antibodies after differentiation into plasma cells, a process that is regulated by Th2 cytokines and CD40/CD40 ligand interaction with a helper T cell. Up to 107 different antibody specificities can be generated in humans by recombination and somatic mutation of the immunoglobulin genes.23 During an immune response, these processes ensure selection of the most appropriate antibodies so that IgG molecules with the highest affinity for antigen are produced. Antibodies generally recognize specific determinants on an antigen that is intact rather than processed or fragmented, and these determinants are termed epitopes; however, unlike T cell epitopes, most B cell epitopes are conformational and are formed from discontinuous regions of the molecule.24 As well as producing antibodies, B cells are important APCs in that they are able to take up antigen via this surface-bound immunoglobulin and enter into cognate recognition of appropriate T cells. This type of antigen presentation may be important in diversifying the T cell response because B cells present multiple epitopes of the antigen to T cells.
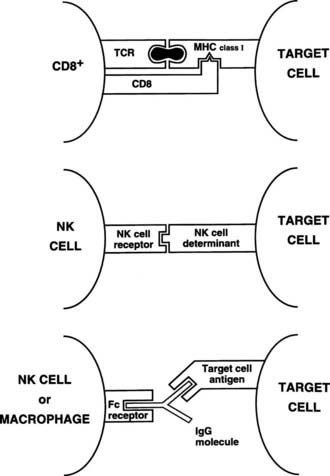
Killer and natural killer cells are CD3 negative and spontaneously destroy target cells with altered surface antigens (particularly reduced MHC class I), such as tumor cells.13 Because killer and natural killer cells express receptors (CD16) for the constant (Fc) region of immunoglobulins, they also can bind to and kill antibody-coated targets, a process called antibody-dependent cell-mediated cytotoxicity (ADCC). This function also can be mediated by macrophages. Thus, although natural killer cells have little antigen specificity, they can be focused on a target via a specific antibody (Fig. 79-5).
SELF-TOLERANCE AND AUTOIMMUNITY
The immune system exists to eliminate foreign antigens but must remain tolerant of (i.e., must not respond to) autoantigens. We now know, particularly through experiments with transgenic mice, that individual mechanisms for ensuring self-tolerance (Table 79-3) are practically never completely successful; a few autoreactive T cells are present in all healthy individuals, although in various states of nonreactivity. Failure of the control mechanisms for ensuring nonreactivity allows clonal expansion of these cells, with subsequent autoimmune disease if the response is sufficiently vigorous.
Table 79-3. Mechanisms for Maintaining Autoreactive T Cell Nonresponsiveness to Target Tissues
| Mechanism | Reversibility |
|---|---|
| Thymic Presentation of Self-Antigens | |
| Deletion | No |
| Anergy | Yes |
| Peripheral Tolerance | |
| Deletion | No |
| Anergy | Yes |
| Clonal ignorance | Yes |
| Immunologic privilege at a target site | Yes |
| Active Suppression | |
| Cytokine network | Yes |
| Idiotype–anti-idiotype networks | Yes |
| Regulatory (Treg) cells | Yes |
During development, the thymus is mainly responsible for eliminating autoreactive T cells (clonal deletion) and for positively selecting appropriate T cells to constitute the immune repertoire.25,26 These processes depend on the interaction of endogenous peptides, presented by thymic APCs, with the naive TCR repertoire. Inevitably, a few T cells escape tolerance, which is particularly likely if specific endogenous antigens are unavailable for presentation in the thymus because of low abundance outside a developing organ, for example. For these antigens, peripheral tolerance, including both deletion and anergy (see Fig. 79-3), may be very important for regulation of self-reactivity.
As well as deletion and anergy, clonal ignorance provides effective protection against autoreactivity. Simply put, autoantigen-specific lymphocytes are harmless unless they become activated by autoantigen; therefore, autoimmune disease will not result from such cells if they do not come into contact with autoantigen, or if the necessary costimulatory signals are not provided. Autoreactive CD8+ T cells and B cells remain harmless or “ignorant” of self-antigens unless activated by helper T cells; therefore, provided that the latter are controlled, autoimmune disease will not result. However, it should be noted that CD8+ T cells and B cells are subject to central tolerance mechanisms in the fetal thymus or bone marrow and liver, respectively.27 Another mechanism for conferring immunologic privilege at an anatomic site, whereby it becomes protected from recognition, is localized expression of FasL, which induces apoptosis in potentially autoaggressive lymphocytes entering the site; examples of such FasL expression are seen in Sertoli cells, corneal cells, and the placental trophoblast.
Subsidiary control over autoreactive T cells is provided by a number of suppressor mechanisms that often are now referred to as regulatory pathways and remain to be fully characterized. Inhibitory cytokines, such as those that produce reciprocal inhibition of Th1 and Th2 subset responses, provide one means of suppression of harmful autoimmune responses,18 but antigen-specific suppressor phenomena have been defined in many situations, especially in animal cell transfer experiments.20,28,29
Tolerance to a self-antigen can be broken at one or more of the levels at which it operates (see Table 79-3) and may involve both genetic and environmental factors:
Experimental Autoimmune Thyroiditis
The development of animal models of experimental autoimmune thyroiditis (EAT) (Table 79-4) has allowed profound insight into the development of thyroid autoimmunity, for example, Fig. 79-6 shows how autoimmune thyroid disease can arise in mice through modulation at the different stages of T cell tolerance, some of the principles of which were described in the preceding section. Several different types of EAT have been described,30 and these types more or less resemble Hashimoto’s thyroiditis, with lymphocytic infiltration of the thyroid, TG antibodies, and variable degrees of hypothyroidism.30 Most recently, attempts to produce animal models of Graves’ disease31,32 have had some success, although they remain incompletely characterized. Key lessons from these animal models can be summarized as follows.
Table 79-4. Summary of the Main Animal Models of Experimental Autoimmune Thyroiditis
| Model | Antigen | Comments |
|---|---|---|
| Immunization usually with adjuvant (mouse, rat, rabbit, guinea pig) | TG, TPO, TSHR | Strain dependent, transient, and transferable via T cells; TSHR does not induce a Graves’ disease–like model because the TSHR antibodies are without stimulatory action |
| Thymectomy induced (mouse, rat) | TG | Depends on time of thymectomy and strain, may require sublethal irradiation; transferable with T cells |
| T cell manipulations (mouse) | TG | Cyclosporine A and transfer of specific T cells to T cell–depleted animals induce thyroiditis |
| Spontaneous (chicken, dog, rat) | TG + other autoantigens | Thyroiditis occurs in OS chickens, beagles, NOD mice, and BB and Buffalo strain rats (NOD and BB animals also have autoimmune diabetes) |
| Virus induced (mouse) | TG + polyendocrine autoantigens | Reovirus infection of certain strains of mice |
| SCID mouse | TG, TPO, TSHR | SCID mice allow long-term study of transplanted thyroid tissue from patients with Graves’ and Hashimoto’s disease; disease does not develop in the mice themselves |
| cDNA Immunization (mouse) | TSHR | Allows production of monoclonal TSHR antibodies |
| Immunization with fibroblasts transfected with TSHR and MHC class II (mouse) | TSHR | Hyperthyroidism and histologic features of Graves’ disease develop, but without lymphocytic infiltration |
MHC, Major histocompatibility complex; SCID, severe combined immunodeficiency; TG, thyroglobulin; TPO, thyroid peroxidase; TSHR, thyroid-stimulating hormone receptor.
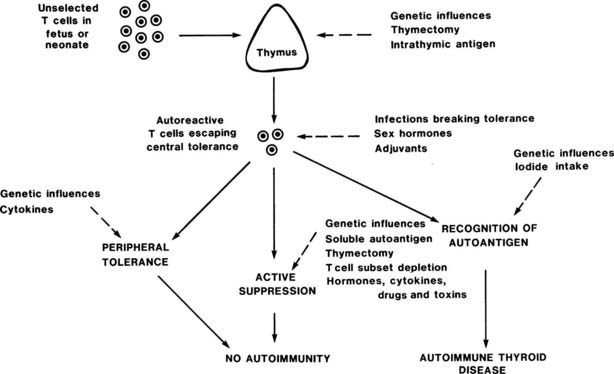
FIGURE 79-6. Control of autoreactive T cells in experimental autoimmune thyroiditis in mice. The sites at which genetic and other factors act are shown as dashed lines; the development of autoimmune thyroid disease depends on the balance between these multiple influences.
(From Weetman AP, McGregor AM: Autoimmune thyroid disease: further developments in our understanding. Endocr Rev 15:788–830.
A strong genetic tendency is apparent in all models such that manipulations that readily induce EAT in one strain may induce no disease or a different autoimmune disease (such as oophoritis or gastritis) in a different strain. In both spontaneous and immunization-induced models, the MHC makes a contribution to genetic susceptibility, but it is clear that other non-MHC loci are also involved; in the OS chicken, for example, these loci control T cell responsiveness, glucocorticoid tonus, and intrinsic properties of the thyroid, which makes the OS chicken more susceptible to autoimmunity.33 Perhaps the most elegant demonstration of susceptibility is the creation of HLA-DRB1*0301 (DR3) transgenic mice; HLA-DR3 is an MHC specificity that is known to confer susceptibility to autoimmune disease in humans (see below). Thyroiditis develops in HLA-DR3 but not HLA-DR2 transgenic mice after TG immunization, thereby confirming that this HLA-DRB1 polymorphism determines, at least in part, susceptibility to autoimmune thyroiditis.34
In addition, a number of environmental and endogenous factors contribute to susceptibility, and more must await discovery. Excess iodine exacerbates spontaneous thyroiditis, possibly through the generation of toxic metabolites that are formed with oxygen in the thyroid, but it is also known that a major T cell epitope on TG requires iodination for recognition by autoreactive T cells.35 Antithyroid drugs suppress EAT with no reduction in thyroid hormone levels, which confirms the direct immunomodulatory effects of these drugs.36
The potential adverse effects of infection are illustrated by the absence of EAT in suitably thymectomized rats that are reared under germ-free conditions; transfer of normal gut microflora will induce disease.37 It is possible that this result is due to the nonspecific effects of gut microflora, such as polyclonal lymphocytic activation or release of cytokines, or to some thyroid cross-reactive antigen. Environmental toxins, such as 3-methylcholanthrene, can induce EAT in genetically susceptible strains of rats.30 Similar to humans, female animals generally have more severe EAT than males do; this difference is dependent on sex hormones; estrogen administration worsens thyroiditis, whereas testosterone has the opposite effect.38
In all models, the role of T cells is paramount, as is illustrated by the effects of T cell manipulation on disease induction (see Table 79-4), and disease is transferred only poorly, if at all, by thyroid antibodies.39 In general, transfer of both CD4+ and CD8+ autoreactive T cells from a donor with EAT is needed to induce EAT in a recipient. A key role for T regulatory cells in preventing EAT has been obvious for decades based on cell transfer experiments.30 It is now established that naturally ocurring CD4+, CD25+ T cells have a partial role in this regard, and additional contributions from antigen-activated T regulatory cells may occur.40 Additional layers of regulation, including cytokine profiles, are likely to be involved, and it is clear from the multiple models of Graves’ disease that a paradigm of TH1/Th2 balance is too simplistic to explain the type of autoimmune response involved.41
No correlation has been found between the level of TG antibodies and the severity of EAT,30 further demonstrating the uncertain pathologic role for these autoantibodies in EAT. Indeed, in transgenic mice, it has been shown that tolerance is not induced in B cells by membrane-bound antigen expressed on thyroid cells, presumably because the preimmune B cells are sequestered from the antigen by basement membrane and endothelium.42 Because tolerance is induced in T cells in this model, it appears that somehow thyroid surface antigens can affect the development of these cells, either by transport to the thymus or by some type of peripheral tolerance. This observation is important and implies that thyroid autoreactive B cells are frequent but remain ignorant; the frequent appearance of thyroid autoantibodies in otherwise healthy populations could be explained by the activation of these B cells if T cell tolerance breaks down or is bypassed.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


