FIGURE 137-1. Pathways of testosterone biosynthesis and action. In men, testosterone biosynthesis occurs almost exclusively in mature Leydig cells by the enzymatic sequences illustrated. Cholesterol originates predominantly by de novo synthesis pathway from acetyl coenzyme A, with luteinizing hormone (LH) regulating the rate-limiting step, the conversion of cholesterol to pregnenolone within mitochondria, while the remaining enzymatic steps occur in smooth endoplasmic reticulum. The Δ5 and Δ4 steroidal pathways are on the left and right, respectively. Testosterone and its androgenic metabolite, dihydrotestosterone, exert biological effects directly through binding to the androgen receptor and indirectly through aromatization of testosterone to estradiol, which allows action via binding to the estrogen receptor (ER). The androgen and ERs are members of the steroid nuclear receptor superfamily, with a highly homologous structure differing mostly in the C-terminal ligand-binding domain. The LH receptor has the structure of a G protein–linked receptor with its characteristic seven-transmembrane-spanning helical regions and a large extracellular domain that binds the LH molecule, which is a dimeric glycoprotein hormone consisting of an α subunit common to other pituitary glycoprotein hormones and a β subunit specific to LH. Most sex steroids bind to sex hormone–binding globulin (SHBG), which binds tightly and carries the majority of testosterone in the bloodstream.
Testicular testosterone secretion is principally governed by luteinizing hormone (LH) through its regulation of the rate-limiting conversion of cholesterol to pregnenolone within Leydig-cell mitochondria by the cytochrome P450 cholesterol side chain–cleavage enzyme complex located on the inner mitochondrial membrane. Cholesterol supply to mitochondrial steroidogenic enzymes is governed by proteins including sterol carrier protein 2.11 This facilitates cytoplasmic transfer of cholesterol to mitochondria, as well as steroidogenic acute regulatory protein (StAR)12 and peripheral benzodiazepine receptor,13 which govern cholesterol transport across the mitochondrial membrane. All subsequent enzymatic steps are located in the Leydig-cell endoplasmic reticulum. The high testicular production rate of testosterone creates both high local concentrations (up to 1 µg/g tissue, ∼100 times higher than blood concentrations) and rapid turnover (200 times per day) of intratesticular testosterone14; however, the precise physical state in which such high concentrations of intratesticular testosterone and related steroids exist in the testis remains to be clarified.
SECRETION
Testosterone is secreted at adult levels during three epochs of male life: transiently during the first trimester of intrauterine life (coinciding with masculine genital tract differentiation), during early neonatal life as the perinatal androgen surge (with still undefined physiologic significance), and continually after puberty to maintain virilization. The dramatic somatic changes of male puberty are the consequence of striking increases in testicular secretion of testosterone rising approximately 30-fold over levels that prevail prior to puberty and in women or castrate men, originating from extratesticular sources. After middle age, there are gradual decreases in circulating testosterone as well as increases in gonadotrophin and sex hormone–binding globulin (SHBG) levels,15–17 with these trends being exaggerated by the coexistence of chronic illness.18–20 These age-related changes, including the effects of concomitant accumulation of chronic disease states, are functionally attributable to impaired hypothalamic regulation of testicular function,21–24 as well as Leydig cell attrition7 and dysfunction25–27 and atherosclerosis of testicular vessels.28 As a result, the aging hypothalamic-pituitary-testicular increasingly operates with progressive multilevel functional defects that in concert lead to reduced circulating testosterone levels during male aging.29
Testosterone, like other lipophilic steroids secreted from steroidogenic tissues, leaves the testis by diffusing down a concentration gradient across cell membranes into the bloodstream, with smaller amounts appearing in the lymphatics and tubular fluid. After puberty, over 95% of circulating testosterone is derived from testicular secretion, with the remainder arising from extragonadal conversion of precursors with low intrinsic androgenic potency such as DHEA and androstenedione. These weak androgens, predominantly originating from the adrenal cortex, constitute a large circulating reservoir of precursors for conversion to bioactive sex steroids in extragonadal tissues including the liver, kidney, muscle, and adipose tissue. Unlike in women, where adrenal androgens are the major source of biologically active androgens, endogenous adrenal androgens contribute negligibly to direct virilization of men,9 and residual circulating and tissue androgens after medical or surgical castration have minimal biological effect on androgen-sensitive prostate cancer.30 Conversely, however, adrenal androgens make a proportionately larger contribution to the much lower circulating testosterone concentrations in children and women (∼5% of men), in whom blood testosterone is derived approximately equally from direct gonadal secretion and indirectly from peripheral interconversion of adrenal androgen precursors. Exogenous DHEA at physiologic replacement doses of 50 mg/day orally31 is incapable of providing adequate blood testosterone for androgen replacement in men but produces dose-dependent increases in circulating estradiol in men32,33 and hyperandrogenism in women.10
Hormone production rates can be calculated from either estimating metabolic clearance rate (from bolus injection or steady-state isotope infusion using high–specific activity tracers) and mean circulating testosterone levels34,35 or by estimation of testicular arteriovenous differences and testicular blood flow rate.36 These methods give consistent estimates of a testosterone production rate of 3 to 10 mg/day using tritiated37,38 or nonradioactive deuterated39 tracers with interconversion rates of approximately 4% to dihydrotestosterone (DHT)38,40 and 0.2% to estradiol41 under the assumption of steady-state conditions (hours to days). These steady-state methods are a simplification that neglects diurnal rhythm,42,43 episodic fluctuation in circulating testosterone levels over shorter periods (minutes to hours) entrained by pulsatile LH secretion,44 and postural influence on hepatic blood flow.37 The major known determinants of testosterone metabolic clearance rate are circulating SHBG concentration,45 diurnal rhythm,39 and postural effects on hepatic blood flow37,39 influenced by genetic38 and environmental39 factors.
TRANSPORT
Testosterone circulates in blood at concentrations greater than its aqueous solubility by binding to circulating plasma proteins. The most important is SHBG, a high-affinity but low-capacity binding protein. Low-affinity binding proteins include albumin, corticosteroid-binding globulin,46 and α1 acid glycoprotein.47 Testosterone binds avidly to circulating SHBG, a homodimer of two glycoprotein subunits, each comprising 373 amino acids with 3 glycosylation sites, 2 N-linked and 1 O-linked, containing a single high-affinity steroid-binding site. The affinity of SHBG for binding testosterone does not change in liver disease,48 but whether it is influenced by other chronic diseases or pregnancy is not known. SHBG is secreted (1) into the circulation by human but not rodent liver, (2) into the seminiferous tubules of the testis by rodent but not human Sertoli cells, where it is known as testicular androgen-binding protein,50 and (3) by placenta, where it may contribute to the rise in blood SHBG during pregnancy.51 Circulating SHBG is secreted into the bloodstream from the liver, and its blood levels (and thereby total testosterone) are prominently influenced by supraphysiologic hormone exposure of the liver, whether by oral administration (via first-pass hepatic effects) or by parenteral injection of high doses. Consequently, blood SHBG concentrations are characteristically decreased (androgens, glucocorticoids) or increased (estrogens, thyroxine), which produce supraphysiologic hepatic exposure to the exogenous hormones. In contrast, endogenous sex steroids and parenteral (non-oral) administration, which maintain physiologic blood hormone concentrations (transdermal, depot implants), have minimal effects on blood SHBG levels. Other modifiers of circulating SHBG levels include up-regulation by acute or chronic liver disease and androgen deficiency and down-regulation by obesity, protein-losing states, and genetic SHBG deficiency.52,53 Under physiologic conditions, 60% to 70% of circulating testosterone is SHBG bound, with most of the remainder bound to lower-affinity, high-capacity binding sites (albumin, α1 acid glycoprotein, corticosteroid binding protein) and 1% to 2% not protein bound.
Transfer of hydrophobic steroids into tissues is presumed to occur passively according to physicochemical partitioning between the hydrophobic protein binding sites on circulating binding proteins, the hydrophilic aqueous extracellular fluid, and the lipophilic cellular plasma membranes. According to the free-hormone hypothesis,54–56 the free (non-protein-bound) fraction of testosterone is the most biologically active, with the loosely protein-bound testosterone constituting a less accessible but mobilizable fraction, and the largest moiety tightly bound to SHBG, constituting only an inactive reservoir. As the free and/or bioavailable fractions would also have enhanced access to sites of testosterone inactivation by degradative metabolism that terminate androgen action, the free fractions may alternatively be considered the most evanescent and least active. Consequently, a theoretical basis for the free-hormone hypothesis is questionable. Furthermore, empirical evidence indicates that rather than being biologically inert, SHBG participates actively in cellular testosterone uptake via specific SHBG membrane receptors, uptake mechanisms, and signaling via G protein and cyclic adenosine monophosphate (cAMP).57–61 Consequently, the biological significance and clinical utility of partitioning circulating testosterone into these derived fractions remains to be firmly established.
MEASUREMENT
Measuring blood testosterone concentration is an important part of the clinical evaluation of androgen status and confirming a clinical and pathologic diagnosis of androgen deficiency. The circulating testosterone concentration is a surrogate measure for whole-body testosterone production rate. However, the reliance on a spot measurement of blood testosterone concentration neglects changes in the whole-body metabolic clearance rate, as well as other factors influencing net androgen effects at tissue levels. These include the efficiency of blood testosterone transfer into adjacent tissues during capillary transit and prereceptor, receptor, and postreceptor factors influencing the testosterone activation, inactivation, and action in that tissue. Circulating testosterone levels are also dynamic and feature distinct circhoral and diurnal rhythms. Circhoral LH pulsatility entrains some pulsatility in blood testosterone levels,24 although the buffering effects of the circulating steroid-binding proteins dampens the pulsatility of blood testosterone concentrations. Diurnal patterns of morning peak testosterone levels and nadir levels in the midafternoon are evident in younger and healthy older men42 but lost in some aging men.43 Consequently, it is conventional practice to standardize testosterone measurements to morning blood samples on at least two different days.
The advent of steroid radioimmunoassay in the 1970s made it feasible to measure blood testosterone concentrations affordably with speed and sensitivity. However, cross-reacting steroids and nonspecific matrix effects are limitations on testosterone immunoassays relative to the high specificity of mass spectrometry–based methods,62 the reference method which, however, has been too costly, slow, and inaccessible for routine clinical use. The steep rise in demand for testosterone measurements in clinical practice led to method simplifications to integrate steroid immunoassays into automated immunoassay platforms. These changes, notably eliminating preparative solvent extraction and introducing bulky nonauthentic tracers, undermine the specificity of unextracted testosterone immunoassays,62 particularly at the low circulating testosterone levels such as in women and children.63 Even at the higher testosterone concentrations in men, commercial testosterone immunoassays demonstrate wide discrepancies.64 New-generation mass spectrometers with higher sensitivity and throughput may overcome these limitations of testosterone immunoassays.
Assays to measure blood free-testosterone levels directly in serum samples have been developed using tracer reference methods of equilibrium dialysis,65,66 ultrafiltration,67,68 or various formulae calculations based on immunoassay measurement of total testosterone and SHBG.69 Similarly, another derived testosterone measure, bioavailable testosterone, is defined as the non-SHBG-bound testosterone (in effect the combination of albumin-bound plus unbound testosterone) and can also be measured directly or calculated by a formula from total testosterone and SHBG and albumin measurements. Some estimates of free testosterone, notably the direct analog assay70,71 and the free testosterone index,72 are invalid for use in men, so that various calculational formulae, based on equilibrium-binding assumptions69 or empirical estimation,73–75 have become widely used; however, the equilibrium binding formulae are inaccurate in large-scale evaluation,75 with predictive inaccuracies arising mainly from discrepancies between testosterone immunoassays.76 Overall, the clinical utility of various derived measures of testosterone arising from the intuitively appealing but unproven free-hormone hypothesis remain to be established. For further details of the management of androgen deficiency see Chapter 140.
METABOLISM
After testicular secretion, a small proportion of testosterone undergoes activation to two bioactive metabolites, estradiol and DHT, whereas the bulk of secreted testosterone undergoes inactivation by hepatic phase I and II metabolism to inactive oxidized and conjugated metabolites for urinary and/or biliary excretion (Fig. 137-2).77
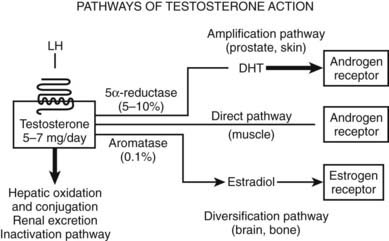
FIGURE 137-2. Pathways of testosterone action. In men, most (>95%) testosterone is produced under luteinizing hormone (LH) stimulation through its specific receptor, a heptahelical G protein–coupled receptor located on the surface membrane of the steroidogenic Leydig cells. The daily production of testosterone (5 to 7 mg) is disposed along one of four major pathways. The direct pathway of testosterone action is characteristic of skeletal muscle, in which testosterone itself binds to and activates the androgen receptor. In such tissues, there is little metabolism of testosterone to biologically active metabolites. The amplification pathway is characteristic of the prostate and hair follicle, in which testosterone is converted by the type 2 5α-reductase enzyme into the more potent androgen dihydrotestosterone (DHT). This pathway produces local tissue-based enhancement of androgen action in specific tissues according to where this pathway is operative. The local amplification mechanism was the basis for the development of prostate-selective inhibitors of androgen action via 5α-reductase inhibition, the forerunner being finasteride. The diversification pathway of testosterone action allows testosterone to modulate its biological effects via estrogenic effects that often differ from androgen receptor–mediated effects. The diversification pathway, characteristic of bone and brain, involves the conversion of testosterone to estradiol by the enzyme aromatase, which then interacts with the estrogen receptors α and/or β. Finally, the inactivation pathway occurs mainly in the liver, with oxidation and conjugation to biologically inactive metabolites that are excreted by the liver into the bile and by the kidney into the urine.
The amplification pathway converts approximately 4% of circulating testosterone to the more potent pure androgen, DHT.38,40 Relative to testosterone, DHT has higher binding affinity78 and 3 to 10 times greater molar potency in transactivation79–81 of the AR. Testosterone is converted to the most potent natural androgen, DHT, by the 5α-reductase enzyme that originates from two distinct genes (I and II).82 Type 1 5α-reductase is expressed in the liver, kidney, skin, and brain, whereas type 2 5α-reductase is characteristically expressed strongly in the prostate but also at lower levels in the skin (hair follicles) and liver.82 Congenital 5α-reductase deficiency due to mutation of the type 2 enzyme protein83 leads to a distinctive form of genital ambiguity causing undermasculinization of genetic males, who may be raised as females but in whom puberty leads to marked virilization, including phallic growth, normal testis development and spermatogenesis,84 and bone density,85 as well as, occasionally, masculine gender reorientation.86 Prostate development remains rudimentary,87 and sparse body hair without balding is characteristic.88 This remarkable natural history reflects the dependence of urogenital sinus–derivative tissues on strong expression of 5α-reductase as a local androgen amplification mechanism for their full development. This amplification mechanism for androgen action was exploited in developing azasteroid 5α-reductase inhibitors.89 Because the type 2 5α-reductase enzyme results in over 95% of testosterone entering the prostate being converted to the more potent androgen, DHT,90 blockade of that isoenzyme (the expression of which is largely restricted to the prostate) confines the inhibition of testosterone action to the prostate (and other urogenital-sinus tissue derivatives) without blocking extraprostatic androgen action. DHT circulates at approximately 10% of blood testosterone concentrations, owing to spillover from the prostate91,92 and nonprostatic sources.93 Whereas genetic mutations disrupting type 2 5α-reductase produce disorders of urogenital sinus–derived tissues in men and mice,94 genetic inactivation of type 1 5α-reductase has no male phenotype in mice, but mutations of the human type 1 enzyme have not been reported. An important issue is whether eliminating intraprostatic androgen amplification by inhibition of 5α-reductase can prevent prostate disease. A major 10-year chemoprevention study randomizing nearly 19,000 men over 55 years of age without known prostate disease to daily treatment with an oral 5α-reductase inhibitor, finasteride, or placebo observed a cumulative 25% reduction after 7 years of treatment in early stage, organ-confined, low-grade prostate cancer.95 Although the study was not designed to determine survival benefit, there was an apparent stage shift toward higher grade but still organ-confined cancers, possibly a medication effect on prostate and tumor histology.96 A further placebo-controlled study of 8000 men older than 50 years of age at high risk of prostate cancer using a dual (type 1 & 2) 5α-reductase inhibitor, dutasteride, underway97 will clarify these findings that highlight the importance of androgen amplification within the prostate in the origin of cancer during the long latent premalignant phase. Whether or not preventive use of prostatic 5α-reductase inhibition in men with high prostate cancer risk proves warranted, novel synthetic androgens refractory to 5α-reductive amplification may have advantages for clinical development.
The diversification pathway of androgen action involves testosterone being converted by the enzyme aromatase to estradiol98 to activate estrogen receptors (ERs) (see Fig. 137-2). Although this involves only a small proportion (∼0.2%) of testosterone output, the higher molar potency of estradiol (∼100-fold higher versus testosterone) makes aromatization a potentially important mechanism to diversify androgen action via ER-mediated effects in tissues where aromatase is expressed. The diversification pathway is governed by the cytochrome P450 enzyme (CYP19) aromatase.98,99 In eugonadal men, most (∼80%) circulating estradiol is derived from extratesticular aromatization.41 The biological importance of aromatization in male physiology was first recognized in the early 1970s100 when the local conversion of testosterone to estradiol within the neural tissues was identified and subsequently shown to have an important role in mediating testosterone action, including negative feedback as well as activational and organizational effects on the brain.101 More recently, the importance of local aromatization in testosterone action has been reinforced by the striking developmental defects in bone and other tissues of men and mice with genetic inactivation of aromatase leading to complete estrogen deficiency due to genetic inactivation of the aromatase.102 This phenotype is also strikingly similar to that of a man103 and mice103 with genetic mutations inactivating ERα. Furthermore, men with aromatase deficiency treated with exogenous estradiol or other estrogens also demonstrated significant bone maturation. By contrast, genetic inactivation of ERβ has no effect on male mice,104 and no human mutations have been reported. Aromatase expression in tissue such as bone105 and brain101 may influence development and function by variation in aromatization that modulates local tissue-specific androgen action. By contrast, other tissues like mature liver and muscle express little or no aromatase. Nevertheless, despite the importance of aromatization for male bone physiology, other observations indicate that androgens acting via ARs have important additional direct effects on bone. These include the greater mass of bone in men despite very low circulating estradiol concentrations compared with young women,106 the failure of androgen-insensitive rats lacking functional ARs but normal estradiol and ERs to maintain bone mass of normal males,107 and the ability of nonaromatizable androgens to increase bone mass in estrogen-deficient women.108,109 Testosterone action on bone and in the brain are not accounted for solely as a prohormone for local estradiol production (and action via ERs α and/or β), and AR-mediated effects are required to manifest the full spectrum of testosterone effects on bone110,111 and in the brain.112 Further studies are needed to fully understand the significance of aromatization in maintaining androgen action in mature male animals.113
Testosterone is metabolized to inactive metabolites in the liver, kidney, gut, muscle, and adipose tissue. Inactivation is predominantly by hepatic oxidases (phase I metabolism), notably the cytochrome P450 3A family,114 leading ultimately to oxidation of most oxygen moieties followed by hepatic conjugation to glucuronides (phase II metabolism), which are rendered sufficiently hydrophilic for renal excretion. Uridine diphospho (UDP)-glucuronosyltransferase (UGT) enzymes UGT2B7, UGT2B15, and UGT2B17 catalyze most phase II metabolism (glucuronidation) of testosterone with, 2B17 being quantitatively the most important.115 A functional polymorphism of UGT2B17, a deletion mutation several times more frequent in Asian than European populations,116 explains the concordant population difference in testosterone to epitestosterone (T/E) ratio,116 a World AntiDoping Agency–approved urine screening test for testosterone doping in sport, which may constitute ethnic-differential false negatives in surveillance for exogenous testosterone doping.117
The metabolic clearance rate of testosterone is reduced by increases in circulating SHBG levels45 or decreases in hepatic blood flow (e.g., posture)37 or function. Theoretically, drugs that influence hepatic oxidase activity could alter metabolic inactivation of testosterone, but empirical examples of sufficient magnitude to influence clinical practice are rare. Rapid hepatic metabolic inactivation of testosterone leads to both low oral bioavailability118,119 and short duration of action when injected parenterally.120 To achieve sustained androgen replacement, these limitations dictate the need to deliver testosterone via parenteral depot products (e.g., injectable testosterone esters, testosterone implants, transdermal testosterone) or oral delivery systems that either bypass hepatic portal absorption (buccal,121,122 sublingual,121,123 gut lymphatic124) or use synthetic androgens with substituents rendering them resistant to first-pass hepatic inactivation.125
REGULATION
During sexual differentiation in early intrauterine life, the testosterone required for masculine sexual differentiation is secreted by fetal Leydig cells. The regulation of this fetal Leydig-cell testosterone secretion appears to differ between species. Higher primate and equine placenta secrete a chorionic gonadotropin during early fetal life126 that may drive fetal human Leydig-cell steroidogenesis127 at the relevant time. By contrast, in subprimate mammals, male sexual differentiation occurs without expression of any placental gonadotropin and prior to the time when pituitary gonadotropin secretion starts, so that fetal Leydig-cell testosterone secretion may be autonomous of gonadotropin stimulation during fetal development of most mammalian species.128
Puberty is initiated by a still mysterious suprahypothalamic process (involving a developmental clock and multiple permissive processes129) that lifts the central neuroendocrine restraint on the final common pathway that drives reproductive function in the mature male, episodic secretion of gonadotropin-releasing hormone (GnRH) from hypothalamic neurons.44 Various explanatory theories including the gonadostat, somatometer,130 neurally-driven changes in GABAergic inhibition and glutaminergic stimulation,131 and triggering by kisspeptin-1 secretion and activating its receptor GPR54132,133 are proposed to explain the restraint and resurgence of the hypothalamic GnRH pulse generator, without a comprehensive picture having yet emerged. Hypothalamic GnRH neurons are functional at birth, but after the perinatal androgen surge remain tonically suppressed during infantile life. Puberty is initiated by a maturation process that awakens the dormant hypothalamic GnRH neurons to unleash mature circhoral patterns of pulsatile GnRH secretion, which in turn entrains pulsatile LH secretion from pituitary gonadotropes. Initially this resurgence of pulsatile GnRH and LH secretion occurs mainly during sleep134 but eventually extends throughout the day with a persisting underlying diurnal rhythm. The timing and tempo of male puberty is under tight genetic control, encompassing nutrition influences on body weight and composition,135 with a correspondingly growing number of genetic causes of delayed puberty identified.136 Environmental factors that optimize growth (e.g., higher socioeconomic status with better nutrition and health care) may explain secular trends to earlier puberty with increased statural growth,137,138 whereas claims that exposure to hormonally active chemical pollution contributes to earlier puberty139 remain speculative.140
After birth, testicular testosterone output is primarily regulated by the pulsatile pattern of pituitary LH secretion. This is driven by the episodic secretion of GnRH from hypothalamic neurons into the pituitary portal bloodstream, providing a direct short-circuit route to pituitary gonadotropes. Under this regular but intermittent GnRH stimulation, pituitary gonadotrophs secrete LH in high amplitude pulses at approximately 60- to 90-minute intervals with minimal intervening LH secretion between pulses, with the net effect that circulating LH levels are distinctly pulsatile.141 This pulsatile pattern of trophic hormone exposure maintains Leydig-cell sensitivity to LH to maintain mature male patterns of testicular testosterone secretion.
LH stimulates Leydig-cell steroidogenesis via increasing substrate (cholesterol) availability and activating rate-limiting steroidogenic enzyme and cholesterol transport proteins. LH is a dimeric glycoprotein consisting of an α subunit common to the other glycoprotein hormones—human chorionic gonadotropin (hCG), follicle-stimulating hormone (FSH), and thyrotropin-stimulating hormone (TSH)—and a β subunit providing distinctive biological specificity for each dimeric glycoprotein hormone by dictating its specific binding to the LH/hCG rather than the FSH or TSH receptors142,143 These cell-surface receptors are highly homologous members of the heptahelical, G protein–linked family of membrane receptors. LH receptors are located on Leydig-cell surface membranes and use signal transduction mechanisms involving primarily cAMP and calcium as second messengers to cause protein kinase–dependent protein phosphorylation and DNA transcription, ultimately resulting in testosterone secretion.144 Functionally, hCG is a natural, long-acting analogue of LH because they both bind to the same LH/hCG receptor, and their β subunits are nearly identical, except that hCG has a C-terminal extension of 31 amino acids containing four O-linked, sialic acid–capped carbohydrate side chains. These glycosylation differences confer greater resistance to degradation, which prolongs circulating residence time and biological activity compared with LH,145,146 a feature that has been exploited to engineer longer-acting analogs of other circulating hormones such as FSH,147 TSH,148 and erythropoietin.149
Additional fine tuning of Leydig-cell testosterone secretion is provided by paracrine factors originating within the testis.150 These include cytokines, inhibin, activin, follistatin, prostaglandins E2 and F2α, insulin-like and other growth factors, as well as uncharacterized factors secreted by Sertoli cells. LH also influences testicular vascular physiology by stimulating Leydig-cell secretion of vasoactive and vascular growth factors.151
Testosterone is a key element in the negative testicular feedback cycle through its inhibition of hypothalamic GnRH and, consequently, pituitary gonadotropin secretion. Such negative feedback involves both testosterone effects via ARs and aromatization to estradiol within the hypothalamus.152,153 These culminate in reduction of GnRH pulse frequency in the hypothalamus together with reductions in amplitude of LH pulses due to both reduced GnRH quantal secretion and gonadotropin response to GnRH stimulation. By contrast, the small proportion of estradiol in the bloodstream that is directly secreted from the testes (∼20%) means that circulating estradiol is under minimal physiologic regulation and unlikely to be a major influence on negative-feedback regulation of physiologic gonadotropin secretion in men.
ACTION
Androgen action involves prereceptor, receptor, and postreceptor mechanisms that are centered on the binding of testosterone (or an analog) to the AR. Testosterone undergoes prereceptor activation by conversion to potent bioactive metabolites, DHT and estradiol. The steroidogenic enzyme 5α-reductase has two isozymes, types 1 and 2, which form a local androgen amplification mechanism converting testosterone to the most potent natural androgen, DHT.154 The two isozymes have different chromosomal locations and distinct biochemical features but are homologous genes.82 This local androgen amplification mechanism is exemplified in urogenital sinus–derived tissues, notably external and internal genitalia and the prostate, which characteristically express high levels of 5α-reductase type 2.82 Other tissues such as nongenital skin and liver express 5α-reductase type 1. The other form of prereceptor androgen activation is conversion of testosterone to estradiol by the enzyme aromatase155 which diversifies androgen action by facilitating effects mediated via ERs. Consequently, while DHT may be considered a pure androgen because its bioactivity is solely mediated via AR, testosterone has a wider spectrum of action which includes diversification by aromatization and ER mediated effects. These prereceptor mechanisms provide testosterone with a versatile and subtle range of regulatory mechanism prior to receptor mediated effects, depending on the balance between direct AR mediated versus indirect activational effects and/or ER-mediated mechanisms. In addition, individuals156 and tissues157 vary in their androgenic thresholds and dose-response characteristics to testosterone and its bioactive metabolites.
The Androgen Receptor
The AR is required for masculine sexual differentiation and sexual maturation that ultimately leads to development of a mature testis capable of supporting spermatogenesis and testosterone production that form the basis for male fertility. The human AR is specified by a single X chromosome–encoded gene located at Xq11-12 that specifies a protein of 919 amino acids,1 a classical member of the large nuclear receptor superfamily158 which includes receptors for the five mammalian steroid classes (androgen, estrogen, progesterone, glucocorticoid, mineralocorticoid) as well as for thyroid hormones, retinoic acid, and vitamin D in addition to numerous orphan receptors where the ligand was originally not identified.159 Androgen-receptor expression is not confined to reproductive tissues, and it is ubiquitously expressed, although levels of expression and androgen sensitivity of nonreproductive tissues vary.
The androgen receptor gene has 8 exons specifying a protein of 919 amino acids with the characteristic structure of mammalian steroid receptors. It has an N-terminal domain (NTD) that specifies a long transactivating functional domain (exon 1), a middle region specifying a DNA-binding domain (DBD) consisting of two zinc fingers (exons 2 and 3) separated by a hinge region from the C-terminal ligand binding domain (LBD), which specifies the steroid binding pocket (exons 4 to 8).
The NTD (exon 1) is relatively long, comprising over half (535/919) the overall length of the AR. It has the least conserved sequence compared with other steroid receptors, with a flexible and mobile tertiary structure harboring a transactivation domain (AF-1) that interacts with AR coregulator proteins and target genes.160 Its loose, naturally disordered structure161 also contains three homopolymeric repeat sequences (glutamine, glycine, proline), the most important being the CAG triplet (glutamine) repeat polymorphism.162 The less variable glycine (usually 24 residues) and proline (9 residues) repeat polymorphisms have little apparent independent pathophysiologic significance, although linkage dysequilibrium between the glutamine and glycine repeat polymorphisms requires further haplotype analysis.162 Among healthy people, where the glutamine repeat polymorphism has alleles of lengths between 5 and 35 (population mean ∼ 21), the length of the glutamine repeat is inversely proportional to AR transcriptional efficiency, so that this polymorphism dictates genetic differences between individuals in the androgen sensitivity of their target tissues.163 This genetic variation in tissue androgen sensitivity, though modest in magnitude, influences physiologic responses to endogenous testosterone in prostate size164 and erythropoeisis165 in carefully controlled studies. Wider epidemiologic implications of population variation in the genetic androgen sensitivity as specified by the polyglutamine repeat have been studied in a variety of potentially androgen-sensitive disorders (reviewed in Ref. 162) that include reproductive health disorders and hormone-dependent cancers in men and women, as well as nongonadal disorders where there are significant gender disparities in prevalence. In men, these include prostate166 and other male-preponderant cancers (liver, gastrointestinal, head and neck), prostate hypertrophy, cryptorchidism and hypospadias, and male infertility.167 In women, they include reproductive health disorders (polycystic ovary syndrome, premature ovarian failure, endometriosis, uterine leiomyoma, preeclampsia) and hormone-dependent cancers (breast, ovary, uterus). In addition, studies have also examined risks of obesity, cardiovascular disease, and mental and behavioral disorders (dementia, psychosis, migraine, and personality disorders).162 However, as in many large-scale genetic-association studies,168 the findings remain mostly inconsistent, reflecting methodological limitations—notably in recruitment, participation, and publication bias—as well as multiple hypothesis testing, which tend to inflate spurious associations.
Remarkably, the pathologic elongation of the polyglutamine (CAG triplet) repeat to lengths of over 37 cause a neurodegenerative disease, Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s syndrome), a form of late-onset, slow-progressing but ultimately fatal motor neuron disease,169 one of several late-onset neurodegenerative polyglutamine-repeat disorders.170 Although the extreme length of the polyglutamine repeat does determine mild androgen resistance, these men usually have normal reproductive function in fertility and virilization prior to disease diagnosis in midlife.171 Furthermore, since complete androgen-receptor inactivation in humans and other mammals does not cause motor neuron disease, and female carriers are protected from symptomatic neurodegeneration, SBMA, like other genetic polyglutamine repeat neurodegenerative diseases,172 represents a toxic gain-of-function involving pathologic protein aggregates of the mutant AR.173 Transgenic mouse models of SBMA suggest that testosterone deprivation may slow progression of neuropathy,173 but this hypothesis remains to be tested in humans.
The DBD (exons 2 and 3) consists of approximately 70 amino acids with a high proportion of basic amino acids, including eight cysteines distributed as two sets of four cysteines each forming a zinc coordination center for a single zinc atom thereby creating two zinc fingers. The DBD is highly conserved between steroid receptors reflecting its tightly defined function of forming the two zinc fingers that bind to DNA by intercalating between its grooves. The first zinc finger (exon 2) is directly involved with the major DNA groove of the androgen response element through a proximal (P-box) region whereas the second zinc finger (exon 3) is also responsible for enhancing receptor dimerization through its distal (D-box) region.
The hinge region (first half of exon 4) of approximately 40 amino acids between the DBD and LBD is considered a flexible linker region but may have additional functions involving interactions with DNA (nuclear localization, androgen response element) and protein (AR dimerization, coregulators) which influence AR transcriptional activity.
The LBD (mid–exon 4 to 8) of AR comprises around 250 amino acids which specify a steroid binding pocket, which creates the characteristic high-affinity, stable, and selective binding of testosterone, DHT, and synthetic androgens. While the LBD’s overall architecture is broadly conserved among nuclear receptors, the AR sequence diverges significantly to ensure the specificity of binding from other steroid classes and their different cognate ligands. Structural studies of the AR’s LBD shows it has similar tertiary conformation as other steroid receptors (most closely resembling PR), with 12 stretches of α helix interspersed with short β-pleated sheets. The most C-terminal helix 12 seals the binding pocket and influences whether a bound ligand acts as an agonist or antagonist, as well as forming a hydrophobic surface for binding of coregulator proteins that modify transcriptional activity of the androgen target genes. The LBD also participates in receptor dimerization, nuclear localization, and transactivation via its activation function (AF-2) domain.
The AR has a predominantly nuclear location in androgen target cells, regardless of whether bound to its ligand or not, unlike other steroid receptors which are more often evenly distributed between cytoplasm and nucleus when not bound to their cognate ligands. Androgen binding to the C-terminal LBD causes a conformational change in the AR protein and dimerization to facilitate binding of the ligand-loaded receptor to segments of DNA featuring a characteristic palindromic motif known as an androgen-response element, located in the promoter regions of androgen target genes. Ligand binding leads to shedding of heat shock proteins 70 and 90 that act as a molecular chaperone for the unliganded AR.174 Specific binding of the dimerized, ligand-bound AR complex to tandem androgen-response elements initiates gene transcription, so that the AR acts as a ligand-activated transcription factor. Androgen receptor transcriptional activation is governed by a large number of coregulators175,176 whose tissue distribution and modulation of androgen action remain incompletely understood.
ANDROGEN INSENSITIVITY
Mutations in the AR are relatively common, with over 500 different mutations recorded by 2004177 in the McGill database (http://androgendb.mcgill.ca/), making androgen insensitivity the most frequent form of genetic hormone resistance. Since the AR is an X-chromosomal gene, functionally significant AR mutations are effectively expressed in all affected males, because they are hemizygous. By contrast, women bearing these mutations (including the obligate heterozygote mothers of affected males) are silent carriers without any overt phenotype, because they have a balancing allele and their circulating testosterone levels never rise to postpubertal male levels sufficient to activate AR-mediated effects.
AR mutations produce a very wide spectrum of effects from functionally silent polymorphisms to androgen insensitivity syndromes that display phenotypes proportionate to the impairment of AR function and thereby the degree of deficit in androgen action.1 These clinical manifestations extend from a complete androgen insensitivity syndrome (CAIS, formerly known as testicular feminization) which produces a well-developed female external phenotype in a spectrum spanning across all grades of undervirilized male phenotype to, at the other extreme, a virtually normal male phenotype. The severity of androgen insensitivity can be categorized most simply as complete, partial, and mild, although a more detailed, seven-stage Quigley classification based on degree of hypospadias, phallic development, labioscrotal fusion, and public/axillary hair is also described.1,162 The degree of urogenital sinus–derivative development together with testis descent provide clinical clues to the degree of androgen sensitivity.
CAIS due to completely inactivating AR mutations results in a 46,XY individual with a hormonally active testis that secretes abundant testosterone but which cannot activate AR-mediated action, so no male internal or external genitalia or somatic features develop. However, testosterone aromatization to estradiol is unimpeded, leading to the development of normal female somatic features, including breast and external genital development after puberty. The population prevalence of CAIS is estimated to be at least 1 : 20,000 male births or 1% to 2% among female infants with inguinal hernia.1 The typical presentation of CAIS is a relatively tall, normally developed girl with delayed puberty and/or primary amenorrhea. The clinical features usually include well-developed breasts, hips, and female fat pattern deposition, acne-free facial complexion, and minimal axillary and pubic hair, with testes located within an inguinal hernia or in the abdominal cavity. The uterus and fallopian tubes are absent, and the vagina is short and blind ending, reflecting unimpeded effects of testicular AMH secretion causing regression of müllerian structures that include the upper third of the vagina. Earlier diagnosis is increasingly possible where a prenatal 46,XY karyotype is discrepant from a female phenotype on ultrasound or at birth or among female infants presenting with inguinal hernia.178 The family history may be informative, with infertile maternal (but not paternal) aunts consistent with an X-linked inheritance. Laboratory investigations of postpubertal individuals show elevated blood LH, SHBG (at adult female levels), and testosterone (at adult male levels) prior to gonadectomy. The androgen-sensitivity index, the product of LH and testosterone concentrations, is elevated.179 These features reflect high amplitude and frequency LH pulses due to the absence of effective negative androgenic feedback on the hypothalamus and the increased LH drive to maintain high-normal male levels of testicular testosterone secretion. In untreated individuals, failure to suppress blood SHBG with short-term, high dose androgen administration may be useful confirmation of androgen resistance.180,181 After gonadectomy, blood LH and FSH increase to castrate levels but are partially suppressed by estradiol replacement therapy.
Long-term management includes (1) reinforcing female gender identity with counseling to cope with eventual infertility and acceptance of the genetic diagnosis, (2) postpubertal gonadectomy to prevent the risk of gonadoblastoma (especially if the gonad is impalpable) but allow the completion of puberty, balanced against the low risks of tumor at that age and of unwanted virilization due to any residual AR function or mosaicism,182 and (3) post-gonadectomy estrogen replacement therapy to maintain bone density, breast development, and quality of life. Long-term bone density is often subnormal for age, due not only to the deficit in androgen action but also inadequate post-gonadectomy estrogen replacement, often resulting from suboptimal adherence to medication.85,183–185 Although the long-term outcomes for AR mutations based on large prospective studies of a consistent management approach remain very limited, the clinical outcomes for individuals with CAIS reared as females are reported as successful,186,187 although some gender role and psychosexual functional outcomes remain suboptimal.188,189
Partial androgen insensitivity syndrome (PAIS) is characterized by a full range of external genital virilization and breast development from female to male phenotype, reflecting the functional severity of the AR mutation. A simple clinical guide to the severity of the deficit in AR function is provided by the level of testis descent and phallic development. PAIS was originally recognized under a variety of eponymously named syndromes (Reifenstein, Gilbert-Dreyfus, Lubs, Rosewater) and only more recently clearly distinguished from other developmental disorders of 46,XY individuals with incomplete virilization, especially those due to steroidogenic enzyme defects. Severe forms of PAIS with minimal AR function produce a predominantly female phenotype with clitoromegaly, whereas PAIS with mutations displaying more functional AR are characterized by a male phenotype with various grades of labioscrotal formation (varying from minimal posterior partial labial fusion to labioscrotal fusion and bifid, rugose scrotum) and hypospadias (urinary orifice ranging from perineal aperture to hypospadia with meatus at locations along penile shaft to the corona), micropenis, and gynecomastia, each in inverse proportion to the AR function. These features have been combined into an External Masculinization Score (EMS) ranging from 0 (female) to 12 (male) based on degree of scrotal fusion, phallic development, location of urethral meatus, and testis descent, each scored 0 to 3.190 The biochemical findings in PAIS are similar to those of CAIS but with a wide spectrum of severity from mildly virilized, predominantly female to an undervirilized male phenotype. The increase in blood LH and testosterone are less severe and consistent, but the androgen-sensitivity index179 may help confirm the diagnosis of androgen resistance. Unlike CAIS, which usually presents during adolescence with failure of puberty, PAIS usually presents at birth with ambiguous genitalia requiring a crucial and decisive clinical judgment on sex of rearing to be made rapidly. The expert pediatric endocrinologist must balance the need for early genital surgery and vicarious decision making against the risk of possible subsequent regret by the affected individual as an adult. This makes for inevitably complex, difficult, and contentious choices because the available systematic prospective evidence from long-term follow-up of sex or rearing is still limited. Most intersex individuals due to PAIS, especially those with an EMS of 4 or more,191 are raised as males.190 Genital surgery for hypospadias is often required, and usually uncertainty remains about the adequacy of the potential for postpubertal virilization due to either endogenous or exogenous testosterone. If pubertal progression is inadequate, exogenous testosterone may be useful, but higher than usual dosage may be required to get satisfactory effects. Long-term follow-up of PAIS individuals raised as males has shown apparently adequate psychosexual function despite phallic underdevelopment, limited somatic virilization, and dissatisfaction with outcomes by some patients as adults.192 For those to be reared as females, the management is similar to that for CAIS and involves early genital surgery and prepubertal gonadectomy to prevent unwanted virilization.
Mild androgen insensitivity (MAIS) is the most minor form of androgen insensitivity, displaying near-normal male phenotype with only subtle changes in hair patterns relative to family norms (less body and facial hair, absence of temporal recession or balding) and/or minor defects restricted to spermatogenesis alone. The blood LH and testosterone concentrations are usually but not always consistently elevated, although the androgen-sensitivity index is more consistently raised. In common with mutations in many other genes, making a clear distinction between the most minor grades of clinical pathology and a silent, functionally insignificant polymorphism is challenging and depends on reproducing experimentally the functional consequences of the mutation in an authentic biological system. Ideally, such verification is performed in vivo (e.g., in genetically modified mouse models), but because this is laborious and expensive, it is rarely undertaken. The functional verification of putative mutations is usually undertaken by either in silico prediction of functional effects of structural protein changes from sequence data or in vitro studies of cultured cells or cell-free systems aiming to characterize protein functions. Nevertheless, although informative, the biological fidelity of these surrogate endpoints relative to the in vivo effects on androgen action may remain questionable.
All types of mutations have been reported in the AR gene, including disruption of the reading frame by deletions, insertions, splice-site interruption, and frameshift, which usually produce major interference with function as well as the more common single base substitutions with effects ranging from nil to complete functional inactivation. In addition, mutation can produce less common mechanisms of interrupting AR function, such as inefficient translation, unstable protein, or aberrant translational start sites, all leading to reduced expression of functional AR protein. Mutations occur throughout the AR gene, probably at random; however, those reported are distributed unevenly because the most important functional regions of the gene are sensitive to even minor changes in sequence, whereas the more variable regions may tolerate sequence changes without functional consequences. Over 90% of known mutations are single base substitutions which have pathophysiologic consequences when they change the amino acid sequence in the functionally critical DBD or LBD regions, whereas sequence changes in other regions may not alter AR function, thereby constituting silent polymorphisms. For example, despite forming more than half the AR sequence, few functionally important mutations are reported in the NTD (exon 1). Those described in exon 1 mostly represent major disruptions of the AR protein due to creation of a premature stop codon, a major deletion, or frameshift mutation causing mistranslation onward from exon 1, whereas point mutations are more likely to constitute functionally insignificant (silent) polymorphisms. Mutations in the LBD, comprising some 25% of AR sequence, constitute the majority (∼60%) of reported mutations, whereas mutations in the DBD, representing around 7% of AR sequence, constitute about 14% of cases.177 The functional effects of these two types of mutations generally differ in that LBD mutations demonstrate various degrees of reduced affinity and/or loosened specificity of ligand binding characteristics, but DBD mutations demonstrate normal ligand binding and reduced or absent receptor binding to DNA. The profusion of AR mutations has created numerous experiments of Nature, with multiple different mutations involving the same amino acid and the physiologic consequences depending generally on how conservative is the amino acid substitution. Nevertheless, there are exceptions to such categorization; mutations in regions other than the DBD or LBD sometimes unexpectedly affect DNA or ligand-binding properties, presumably through physical interaction effects in the tertiary structure of the AR in its three-dimensional topography.
The familial occurrence of androgen insensitivity due to X-linked inheritance of mutated AR makes carrier detection and prenatal genetic diagnosis feasible. A carrier female has a 50% chance of having a child bearing the mutant AR allele so they would be either a carrier female or an affected male, and 50% of her fertile daughters will also be carriers. A specific mutation-detection test needs to be established, usually involving PCR-based genotyping for point mutations, although other mutational mechanisms may require more complex genotyping methods. For prenatal genetic diagnosis, now usually applied to chorionic villus samples, the genetic diagnosis must be rapid, reliable, and efficient. However, accurate genetic counseling relies on a consistent and predictable phenotype for any specific genotype. This is usually but not invariably true for AR mutations, since the clinical manifestations for the same mutation are usually consistent in CAIS with rare exceptions.193 For PAIS, the phenotype may vary even within a single family, carrying significant implications for sex of rearing and/or need for genital surgery; skilled genetic counseling is essential.194 Discrepancies in the fidelity of phenotype within families, or between unrelated individuals bearing the identical mutation, is relatively common in PAIS and may be attributable to somatic mosaicism195 or the effect of modifier genes that influence androgen action such as 5α-reductase.196 An exotic, complex DNA breakage repair slippage mechanism has also been described to produce multiple mutations within a single family.197 Wider population genetic screening for AR mutations is not currently cost effective. Despite diminishing costs for increasingly facile genetic testing, the large number of different mutations featuring diverse mechanisms and variable phenotype—which still mostly predict a normal life expectancy but a diminished quality of life—is difficult to cost or cure.198
Acquired androgen insensitivity during life can arise either through postnatal somatic or germline AR mutations or by nongenetic, nonreceptor mechanisms that hinder androgen action. Among overt cases of androgen insensitivity, approximately 30% are absent in the mother’s germline so must arise as a de novo mutation in the postnatal maternal germline195 or in the fetal germline soon after fertilization.199 Somatic AR mutations, arising de novo postnatally in the stem-cell pool of repopulating cells, are theoretically possible but have not been reported. Somatic AR mutations are relatively common in prostate cancer, usually arising in late stage disease palliatively treated by androgen deprivation. The switch of highly androgen-dependent prostate cancer cells to an androgen-deplete milieu may encourage clonal selection of androgen-insensitive sublines to proliferate in the terminal stage of the disease. Genetic instability of prostate cancer cells may also contribute to this process, although somatic AR mutations are rare in other cancers such as liver200 or breast201 cancer in the absence of androgen deprivation. Somatic AR mutations in prostate cancer cells are responsible for the paradoxical antiandrogen withdrawal syndromes observed with nonsteroidal (flutamide, bicalutamide, nilutamide) or steroidal (cyproterone, megestrol) drugs.202,203 In this state, antiandrogen withdrawal or switchover203 produces remission of worsening disease attributable to the occurrence of a de novo AR mutation in prostate cancer cells which alters ligand specificity, turning the nonsteroidal antiandrogens into AR agonists.204 The LNCaP prostate cell line widely used in cancer cell biology research harbors a mutated AR (T877A) which occurs relatively frequently in prostate cancer metastases and can cause the flutamide withdrawal syndrome.205
Acquired androgen insensitivity may occur without AR mutations by mechanisms such as the nonsteroidal (flutamide, bicalutamide, nilutamide) and steroidal (cyproterone acetate) drugs mentioned earlier or drugs that block part of testosterone activation, such as 5α-reductase inhibitors (finasteride, dutasteride), estrogen antagonists, or aromatase inhibitors. In addition, drugs may have physiologic effects or pharmacologic actions that oppose various steps in androgen action (e.g., LH and FSH suppression by estrogens or progestins) or that cause an increase in circulating SHBG, which may influence testosterone transfer from blood into tissues to produce a functional phenocopy of androgen insensitivity.
Hormonal findings reflecting impeded androgen action may cause acquired androgen insensitivity in various disease states and may be reversible with alleviation of the underlying disease. The disease-related mechanisms impeding androgen action vary, but the most frequent is increase in hepatic SHBG secretion due to the underlying disease and/or its drug treatments that impede androgen action by reducing testosterone transport from blood to tissues as part of its overall reduction in metabolic clearance rate of testosterone. For example, in hyperthyroidism, increased blood LH and testosterone concentrations, with clinical features of androgen deficiency,206 are mediated by increased circulating SHBG due to thyroid hormone-induced hepatic SHBG secretion.207 In hypothyroidism, the reduced blood testosterone and SHBG are rapidly corrected by thyroid hormone replacement therapy.206 In epilepsy, anticonvulsant-induced increase in hepatic SHBG secretion appears to be a common denominator in the near-ubiquitous reproductive endocrine abnormalities in men with epilepsy.208 The relative contributions of impaired tissue transfer of testosterone, reduced testosterone metabolic clearance rate,209 or direct antiandrogenic effects of valproate210 remain to be clarified. A similar mechanism of disease- and/or drug-induced increases in hepatic SHBG secretion may explain apparent acquired androgen insensitivity, often reversible with alleviation of the underlying disease in various other conditions such as gluten enteropathy,211,212 Wilson’s disease,213 relapsed acute intermittent porphyria,214 acute alcoholism,215 chronic liver disease, and transplantation.48,216
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


