FIGURE 139-1. A, An 18-year-old male with isolated hypogonadotropic hypogonadism. Note the normal height, long arms and legs, poor muscular development, and prominent gynecomastia. Although his testes and phallus are small, pubic hair is present. It is very unusual for normal boys to have pubic hair growth before the genitalia have begun to develop. B, A  -year-old boy with underdeveloped genitals, absent pubic hair, and gynecomastia. The normal relationship between genital development and pubic hair growth is preserved, and in the ensuing year, spontaneous testis growth began, indicative of constitutional delay of puberty. C, A 51-year-old man with incomplete gonadotropin deficiency and osteoporosis. He had lifelong hypogonadism, including two infertile marriages. His serum testosterone level was 125 ng/dL, and serum luteinizing hormone and follicle-stimulating hormone levels were in the low normal range. His testes were 10 mL in volume.
-year-old boy with underdeveloped genitals, absent pubic hair, and gynecomastia. The normal relationship between genital development and pubic hair growth is preserved, and in the ensuing year, spontaneous testis growth began, indicative of constitutional delay of puberty. C, A 51-year-old man with incomplete gonadotropin deficiency and osteoporosis. He had lifelong hypogonadism, including two infertile marriages. His serum testosterone level was 125 ng/dL, and serum luteinizing hormone and follicle-stimulating hormone levels were in the low normal range. His testes were 10 mL in volume.
In most cases, the testes have a prepubertal histologic appearance. The seminiferous cords are small and contain immature Sertoli cells that appear as a pseudostratified epithelium. A few gonocytes with clear cytoplasm and a central nucleolus are present in the center of the seminiferous cords. The interstitium is composed of loose connective tissue. Mature Leydig cells are absent, but fibroblast-like precursor cells are found.
Serum testosterone levels in most patients are at the low levels characteristic of prepubertal boys, and serum LH levels are barely detectable, even with highly sensitive two-site assays15,16 (Fig. 139-2). LH secretion is generally apulsatile, but a few low-amplitude fluctuations occur in some patients, which presumably reflect attenuated episodes of gonadotropin-releasing hormone (GnRH) secretion. Prolonged pulsatile administration of GnRH usually induces full pubertal development, thus indicating that most cases of IHH are due to lack of normal stimulation of the pituitary by endogenous GnRH.17 The secretion of other pituitary hormones is normal.18 Growth hormone (GH) secretion is reduced slightly because of the sex steroid deficiency and increases to adult values during treatment with testosterone or hCG.19 Restoration of normal gonadotropin release has been reported in some men with IHH following cessation of treatment, suggesting maturation of the hypothalamic-pituitary-testicular (HPT) axis. In one series, 5 of 50 men with absent or partial puberty maintained normal testosterone levels after cessation of treatment, leading the authors to suggest that periodic, brief discontinuation of therapy may be warranted.20
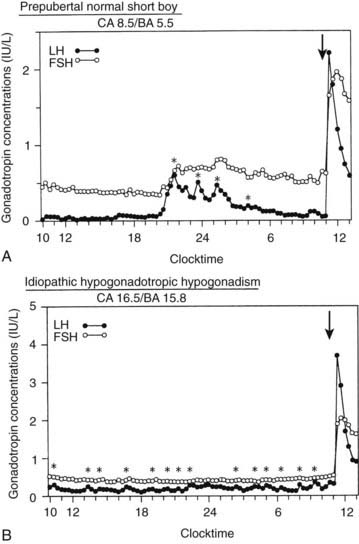
FIGURE 139-2. Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels in blood samples drawn every 20 minutes for 25 hours beginning at 10:00 am in a prepubertal normal boy (A) and an untreated  -year-old with hypogonadotropic hypogonadism (B). The arrows indicate intravenous administration of gonadotropin-releasing hormone at a dose of 25 ng/kg. Plasma LH and FSH levels were measured by DELFIA time-resolved immunofluorometric assays. Asterisks indicate pulses scored by the cluster algorithm. BA, Bone age; CA, chronologic age.
-year-old with hypogonadotropic hypogonadism (B). The arrows indicate intravenous administration of gonadotropin-releasing hormone at a dose of 25 ng/kg. Plasma LH and FSH levels were measured by DELFIA time-resolved immunofluorometric assays. Asterisks indicate pulses scored by the cluster algorithm. BA, Bone age; CA, chronologic age.
(Data from Goji K, Tanikaze S: Comparison between spontaneous gonadotropin concentration profiles and gonadotropin response to low-dose gonadotropin-releasing hormone in prepubertal and early pubertal boys and patients with hypogonadotropic hypogonadism: assessment by using ultrasensitive, time-resolved immunofluorometric assay, Pediatr Res 31:535–539, 1992.)
Other patients have less pronounced clinical signs of hypogonadism and variable testis enlargement, and these signs may be unappreciated until adulthood (see Fig. 139-1C). Basal LH, FSH, and testosterone levels and the gonadotropin response to GnRH stimulation are greater in these men than in men with complete gonadotropin deficiency.21 Spontaneous LH pulses of reduced amplitude and/or frequency often can be detected in the circulation, sometimes with augmentation during sleep.21,22 Because of evidence of testis growth and spermatogenesis, but with incomplete androgenization, the term fertile eunuch has been applied to these men, but partial gonadotropin deficiency is preferred.
Kallmann’s Syndrome
Male hypogonadism with anosmia, first described by Maestre de San Juan in 1856, now is designated as olfactogenital dysplasia to emphasize the association between agenesis of the olfactory bulbs and hypogonadism; the first familial cases were reported by Kallmann and colleagues in 1944.23 Among affected kindreds, the high male-to-female ratio is consistent with an X-linked trait, although kindreds with apparent autosomal dominant or recessive modes of inheritance with variable penetrance also are described.10 Peripheral leukocyte karyotypes generally are normal.
Anosmia or hyposmia is present in 33% to 50% of men with complete IHH and less frequently in patients with partial HH. Magnetic resonance imaging (MRI) reveals that anosmia results from hypoplasia of the olfactory nerves and tracts.24 Genital, somatic, and neurologic abnormalities also may occur25–27 (Table 139-3). Subjects with gonadotropin deficiency and anosmia may have affected relatives without anosmia, thus demonstrating that the clinical features are variable.
Table 139-3. Congenital Defects Associated With Kallmann’s Syndrome
| Neurologic Defects | Genital Defects | Somatic Defects |
|---|---|---|
| Anosmia | Cryptorchidism | Cleft lip |
| Nystagmus | Microphallus | Cleft palate |
| Sensorineural hearing loss | Renal agenesis | |
| Cerebellar ataxia | Horseshoe kidney | |
| Spastic paraplegia | Pes cavus | |
| Learning disability | ||
| Color blindness | ||
| Synkinesis | ||
| Seizures |
Immunocytochemical studies in the fetal mouse have shown that GnRH neurons, together with the central processes of olfactory nerves, migrate from the olfactory placode to the hypothalamus.28 These findings and the awareness that olfactory bulb development depends on contact between central projections of olfactory neurons and forebrain anlage suggest that Kallmann’s syndrome results from a neuron migration defect. Studies of a 19-week stillborn male fetus from a kindred with apparent Kallmann’s syndrome associated with a deletion of the short arm of the X chromosome advanced this hypothesis.29 Whereas GnRH-containing cells were present in the median eminence and preoptic area of age-matched fetuses, no GnRH cells were found in this fetal brain. Instead, dense clusters of GnRH cells were found in his nose, GnRH-containing fibers terminated abruptly on the cribriform plate, and olfactory bulbs and tracts were absent.
Specific Gene Mutations Causing IHH
To date, defects in at least seven genes, specifically, KAL1, FGFR1, GPR54, GnRH receptor, PROK2, and CHD7, have been associated with the IHH phenotype. Mutations in these genes account for about 30% of all cases of Kallmann’s syndrome.
X-Linked HH
Various mutations of the KAL1 gene, found on the short arm of the X chromosome (Xp22.3), have been found in up to 50% of patients with X-linked Kallmann’s syndrome30 (refer to Chapter 122). It is interesting to note that the clinical phenotype varies within families with the same mutation.27,31
Kallmann’s syndrome may be associated with X-linked ichthyosis, which results from steroid sulfatase deficiency (STS gene); this gene is also localized to the pseudoautosomal region of the X chromosome. In these patients, large, dark, dry scales develop on the trunk and limbs because deficient arylsulfatase activity in keratinocytes leads to the accumulation of steroid sulfates. Large Xp deletions may produce a “contiguous gene syndrome” involving the adjacent KAL gene.30
Non–X-Linked IHH
It appears that autosomal gene mutations probably account for most cases of IHH. In one study of 36 familial cases with GnRH deficiency, only 21% could be attributed to X-linkage,26 and when the data were extended to include surrogate markers of IHH (isolated congenital anosmia and delayed puberty), X-linked pedigrees constituted 11%, autosomal recessive 25%, and autosomal dominant 64%. Some of these specific genetic defects have been identified, as is discussed in the following sections. It is possible that digenic mutations may account for some of the phenotypic variability seen in IHH.
FGFR1 Gene
An autosomal dominant variant of HH was identified from the study of patients with contiguous gene syndromes (including Kallmann’s) in which the presence of overlapping deletions at 8p12-p11 led to analysis of the gene encoding fibroblast growth factor receptor-1 (FGFR1).35 In 129 unrelated patients with Kallmann’s, heterozygous FGFR1 mutations were found in four familial and eight sporadic cases, consistent with an autosomal dominant mode of inheritance. One patient born from a consanguineous union had a homozygous missense mutation and was severely affected with cleft palate, agenesis of the corpus callosum, and other defects, leading the authors to suggest that FGFR1 signaling involves the product of the KAL1 gene (anosmin-1) involved in X-linked Kallmann’s. In support of this, some patients with FGFR1 mutations had somatic anomalies typical of X-linked Kallmann’s, such as cleft palate/lip, dental agenesis, and synkinesia. Additionally, both KAL1 and FGFR1 mutations were found in families with Kallmann’s syndrome27 with variable expression of HH within families. However, it has been demonstrated that loss-of-function mutations in FGFR1 may cause nIHH.36
GPR54 Gene
Linkage analysis on chromosome 19p1337 in siblings of a large consanguineous family with HH identified a new autosomal recessive cause of HH involving a 155-bp deletion in GPR54, a G protein–coupled receptor gene that modulates GnRH release. Coincidentally, a role for GPR54 in regulating puberty was proposed after detection of a homozygous mutation of this gene in another consanguineous family38 (refer to Chapter 122).
GnRH Receptor Gene Mutations
The GnRH receptor gene is located on chromosome 4 and encodes a G protein–coupled seven-transmembrane domain receptor.32 Mutations result in autosomal recessive IHH.33 (Refer to Chapter 122.) Impairment of GnRH receptor expression and/or signaling may be complete or partial, the latter allowing some response to GnRH34; the resulting phenotype varies from complete HH (cryptorchidism, microphallus, undetectable gonadotropins) to partial HH with preserved fertility.
Prokineticin 2 and Prokineticin Receptor 2 Genes
Mutations in the genes encoding the G protein–coupled prokineticin receptor 2 (PROKR2) and one of its ligands prokineticin 2 (PROK2) were sought and identified in a cohort with Kallmann’s syndrome following the observation that knockout murine models for both the receptor and the ligand resulted in abnormal olfactory bulb development and sexual maturation. Additional studies revealed a range of reproductive and nonreproductive phenotypes in both Kallmann’s syndrome and normosmic IHH (nIHH) cohorts, including asymptomatic carriers.39
CHD7
CHD7 is a chromatin-remodeling protein that plays a role in puberty. Recently, it has been reported that spontaneous mutations are found in 6% of patients with HH/Kallmann’s syndrome.40
CONGENITAL HYPOGONADISM WITH SYSTEMIC DISORDERS
Neurologic Disorders
In a variety of other syndromes, neurologic abnormalities are associated with hypogonadism (Table 139-4) due to elements of both HH and, in some settings, primary testicular disease. In some of these conditions, patients may not reach adulthood and delayed puberty rather than hypogonadism may be present.
Table 139-4. Clinical Syndromes of Hypogonadism and Neurologic Dysfunction
LEOPARD, Lentigines (multiple), electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, deafness (sensorineural).
The Prader-Willi syndrome (PWS), characterized by neonatal hypotonia, obesity, short stature, small hands and feet, mental retardation, and hypogonadism,41 is diagnosed predominantly in childhood, with most cases being sporadic. Deletions of chromosome 15q of paternal origin are generally present. Subjects often fail to complete puberty spontaneously, although some patients may have early pubarche or, more rarely, precocious puberty. In one study of 42 males, all had cryptorchidism (86% bilateral), and small testes and scrotal hypoplasia are present in 76% and 69%, respectively.42 Total testosterone levels are low but exceed those of prepubertal boys. Basal LH and FSH concentrations are reduced, and the acute response to GnRH is attenuated.43 Treatment with clomiphene may stimulate gonadotropin secretion, as may long-term treatment with GnRH.44 These data suggest suppression of GnRH secretion through an obesity-associated increase in circulating estrogen concentrations, but this mechanism would not explain the findings of cryptorchidism and microphallus, which imply an intrauterine disturbance. A recent study of eight infants and six peripubertal boys suggests a combined hypothalamic and testicular defect.45 Moreover, patients with PWS are GH deficient, with evidence of hypothalamic dysfunction.46 Cryptorchidism may further damage the testes, which would explain the subnormal testosterone response to long-term treatment with hCG. Testosterone treatment is indicated in selected patients.
Patients with the Laurence-Moon-Biedl syndrome have pigmented retinal dystrophy, obesity, mental retardation, polydactyly, renal structural abnormalities, spastic paraparesis, and hypogonadism.47 The designation Bardet-Biedl syndrome often is used in the absence of spastic paraparesis. The reproductive phenotype is variable and includes delayed puberty, microphallus, and, in adulthood, small testes and sterility. Testosterone levels usually are normal, and FSH and LH levels are increased or normal.48 The disorder often is inherited as an autosomal recessive trait, but its cause is unknown.
Congenital oculofacial paralysis (Möbius’ syndrome) is characterized by cranial nerve paralysis and includes eye movement disorders, seizures, mental retardation, gait disturbance, limb anomalies (Poland’s syndrome), and hypogonadism.49 It is suggested to represent a syndrome of rhombencephalic maldevelopment involving motor nuclei and axons, in addition to traversing long tracts, but feeding and respiratory problems and poor motor development suggest a regional developmental disorder.50 Several patients have had documented gonadotropin deficiency.51
Lowe’s syndrome is a rare X-linked disorder characterized by bilateral congenital cataracts, hypotonia, mental retardation, renal tubular acidosis, and hypogonadism. It is caused by loss-of-function mutations of the OCRL gene, encoding an inositol polyphosphate 5-phosphatase,52 which leads to abnormal actin cytoskeleton structure and abnormal formation and function of tight and adherent junctions.53
Noonan’s syndrome features multiple congenital anomalies, including a characteristic facies, valvular heart disease, short stature, and hypogonadism. In one series, 77% of affected males had cryptorchidism.54 Noonan’s syndrome is an autosomal dominant condition affecting 1 in 1000 to 2500 births; however, 50% of cases are sporadic.55 In half the patients, the gene has been mapped to chromosome 1256 and involves missense mutations in the protein-tyrosine-phosphatase, non–receptor type II (PTPNII) gene that encodes a protein called SHP2. This protein is essential in intracellular signaling pathways involved in development. Cryptorchidism is associated with features of primary testicular failure, including hypospermatogenesis and androgen deficiency,57 although a patient with idiopathic hypopituitarism and Noonan’s syndrome was reported.58
The multiple lentigines (LEOPARD) syndrome is an autosomal disorder characterized by lentigines (L), electrocardiographic conduction defects (E), ocular hypertelorism (O), pulmonic stenosis (P), abnormal genitalia (A), retarded growth (R), and deafness (D). The clinical features of LEOPARD syndrome are similar to those of Noonan’s syndrome. Recently, a different mutation in the PTPNII gene was described, leading to the hypothesis that some PTPNII mutations are associated with the typical Noonan phenotype, but that others are associated with both the Noonan phenotype and features such as multiple lentigines or café-au-lait spots.59
Carpenter’s syndrome is an autosomal recessive disorder in which hypogonadism is associated with obesity, acrocephaly, craniosynostosis, and agenesis of the hands and feet.
HH may occur in adrenal hypoplasia congenita (AHC), a rare disorder usually seen in neonates, which generally is fatal if not treated with glucocorticoids and mineralocorticoids,60 although other cases present later in childhood. The severity of the HH phenotype is variable, with cryptorchidism sometimes being present. The DAX1 gene, located on Xp21, is responsible for AHC and encodes a member of the orphan nuclear hormone superfamily.61 DAX1 and a second transcription factor, steroidogenic factor-1 (SF-1), colocalize in the testis, ovary, adrenal cortex, pituitary, and hypothalamus. Null mutations in these genes produce a similar phenotype lacking adrenal glands and gonads, which suggests that the functions of DAX1 and SF-1 are interrelated.62 (Refer to Chapter 122 for additional details.)
Stimulation with pulsatile GnRH usually fails to increase gonadotropin secretion, although responses in some cases suggest an underlying problem with endogenous GnRH production.63 The existence of a primary testicular defect in some cases is suggested by the failure of hCG to induce normal spermatogenesis.34
IDIOPATHIC PARTIAL HYPOPITUITARISM AND PANHYPOPITUITARISM
HH may occur together with other pituitary hormone deficiencies, most commonly GH deficiency.64 The sella turcica is often small, and contrast-enhanced MRI reveals a small pituitary gland, a poorly developed pituitary stalk, and superior dislocation of the neurohypophysis.65 Because of the association with adverse perinatal events, such as breech birth or urgent cesarean birth, trauma to the stalk vasculature has been suggested to cause this syndrome. The presence of micropenis in some patients, however, implies a disturbance that began earlier in gestation, and impaired GH responsiveness to GH-releasing hormone stimulation suggests a hypothalamic disorder. Multiple classes of transcription factors determine the development of the anterior gland. One gene, prophet of pit-1 (Prop-1), is important in the development of Pit-1 lineages, that is, somatotrophs, lactotrophs, and thyrotrophs, as well as gonadotrophs. Thus, patients with combined gonadotropin, GH, prolactin (PRL), and thyroid-stimulating hormone (TSH) deficiency often have Prop-1 gene mutations.66 The same Prop-1 mutation may produce a variable phenotype in terms of hormonal deficiencies and their time of onset, underlining the need for continuous follow-up67 (see Chapters 8 and 16 and 122).
ACQUIRED HYPOGONADOTROPIC HYPOGONADISM
Mass Lesions of the Sella and Suprasellar Region
Gonadotropin deficiency may result from a space-occupying lesion (including hemorrhage) within the sella that compresses and destroys the normal pituitary gland, or from a suprasellar lesion that interrupts the nerve fibers bringing GnRH to the hypophyseal portal circulation. In addition to having hypogonadism, these men often have headaches and visual disturbances, which are characteristic symptoms of a cranial base mass lesion, and variable manifestations of panhypopituitarism. Effects of surgery or external beam radiation therapy to the hypothalamus-pituitary may further damage the endocrine function of the pituitary.68 Pituitary tumors secreting adrenocorticotropic hormone (ACTH) or PRL produce hypogonadism through specific hormonal mechanisms, as is described in the following sections (see Chapter 16).
Cushing’s Syndrome
Decreased libido, erectile dysfunction, and infertility are common complaints among men with Cushing’s syndrome resulting from ACTH-producing pituitary adenomas, adrenal tumors, or tumors outside the pituitary that produce ACTH. Serum testosterone levels usually are low, and basal and GnRH-stimulated LH concentrations frequently are suppressed.69 Increased cortisol production appears to be responsible for the gonadotropin deficiency because adrenalectomy or treatment with mitotane or the glucocorticoid antagonist mifepristone (RU486) restores LH secretion and gonadal function,70 and because glucocorticoids have been shown to decrease transcriptional activity of the mouse GnRH gene.71 The action of glucocorticoids in inducing aromatase activity in fibroblasts72 may contribute to the hypogonadism and gynecomastia. Decreased SHBG levels further lower the total testosterone concentration. Prolonged glucocorticoid treatment of men with otherwise normal testicular function also produces gonadotropin deficiency.73 The combination of hypogonadism and hypercortisolemia leads to loss of muscle mass, weakness, and osteopenia.
Prolactin-Producing Pituitary Adenomas
Men with prolactinomas often were first seen late in the course of their disease with headaches, disturbed vision secondary to enlargement of the pituitary adenoma, and panhypopituitarism.74,75 However, recognition of microprolactinomas is now common because of earlier evaluation of patients with reduced libido and potency, clinical hypogonadism, gynecomastia, or infertility. Teenage boys with delayed pubertal development also should be evaluated for prolactinoma.76 The frequent finding of eunuchoid body proportions in adult men with prolactinomas dates the onset of the tumor to adolescence. In spite of very high levels of prolactin in serum, only about 10% to 20% of men with prolactinomas have galactorrhea,77 presumably because circulating estrogen levels are too low to stimulate mammary gland growth and development.
Serum testosterone levels are low in 50% to 74% and 73% to 93% of men with microprolactinomas and macroprolactinomas, respectively.77,78 Serum LH levels parallel those of testosterone, and the attenuated pulsatile pattern of LH secretion found79 indicates that a decline in LH secretion is responsible for reduced testosterone production. This defect appears to occur in GnRH secretion, because GnRH can restore testicular function to normal.80 In situ hybridization studies in rats with experimental hyperprolactinemia have demonstrated a reduction in GnRH mRNA levels per cell and a resulting decline in GnRH receptor concentration. Reduced semen quality can be present and semen volume low, reflecting reduced androgen action on accessory sex glands.77 Treatment of microprolactinomas and macroprolactinomas with dopamine agonists, bromocriptine and cabergoline, suppresses serum prolactin levels into the normal range in 75% to 83% of microadenomas and macroadenomas,77,78 with a prompt rise in LH and testosterone concentrations, although maximum testosterone levels may not be achieved for several months. Libido and erectile function usually improve,81 as does semen quality.77 Should normalization of prolactin levels fail to occur and serum LH and testosterone production remain low, then testosterone treatment is required. In such patients seeking fertility, gonadotropin therapy is required. The finding that sexual dysfunction may not improve with testosterone replacement alone, whereas coadministration of dopamine agonists and testosterone may increase libido and potency,74 suggests a direct inhibitory action of prolactin on a CNS center controlling sexual arousal.
Miscellaneous Causes of Gonadotropin Deficiency
Infiltrative disorders may affect the hypothalamic-pituitary region. Histiocytosis X may involve the basal hypothalamus, as well as the skeleton, skin, and lungs. Diabetes insipidus and GH deficiency appear to be the most common endocrine disturbances,82 but HH has been reported.83 Sarcoidosis of the hypothalamic-pituitary region is uncommon but may cause anterior pituitary hormone deficiency, diabetes insipidus, and hyperprolactinemia.84 Imaging studies may reveal a solid or cystic sellar or suprasellar mass or pituitary stalk thickening.85
Gonadotropin deficiency due to sex steroid feedback may occur in men with Leydig cell tumors of the testis86 or with benign or malignant tumors of the adrenal cortex that produce estradiol or estrone. Gynecomastia may be the initial complaint in these men and testosterone levels may be reduced,87 requiring diagnostic imaging of the adrenal glands and testicular sonography.
Production of hCG by a choriocarcinoma may increase circulating levels of estrone and estradiol sufficiently to produce gynecomastia and suppress pituitary gonadotropin secretion.88 hCG also stimulates Leydig cell testosterone production, but the testis may be replaced by tumor or damaged by X-irradiation or chemotherapy, so serum testosterone levels are variable.89 Secondary HH also may be induced by sex steroid administration (e.g., anabolic steroid abuse), as is discussed later.
SELECTIVE DEFICIENCY OF LUTEINIZING OR FOLLICLE-STIMULATING HORMONE
Naturally occurring mutations of the gonadotropins and their receptors are rare but provide valuable insights into their structural and functional relationships.90 (Refer to Chapter 122.) Selective deficiency of LH or FSH is exceedingly rare. Biologically inactive LH molecules resulting from missense mutations in LH-β are associated with pubertal failure, androgen deficiency, and infertility.91,92
Several cases of isolated FSH deficiency have been described, including that of an 18-year-old boy with delayed puberty, very small testes, and undetectable serum FSH levels before and after stimulation with GnRH, who unexpectedly had a low serum testosterone and elevated LH, suggesting a concomitant defect of Leydig cell function.93
Gonadotropin receptor mutations are more common than those of the gonadotropins. Men homozygous for an inactivating mutation in the FSH receptor gene were identified by screening male relatives of affected women with primary ovarian failure.94 These men were normally virilized but had small testes, variable sperm concentrations, moderately elevated serum FSH levels, and reduced inhibin B levels, but normal testosterone levels. Activating mutations of the LH receptor gene produce male limited gonadotropin-independent precocious puberty (testotoxicosis) (see Chapter 121), whereas inactivating LH receptor mutations cause Leydig cell hypoplasia with fetal testosterone deficiency, leading to genital ambiguity, as is discussed elsewhere (see Chapter 122).
TREATMENT OF GONADOTROPIN-DEFICIENT MEN
Androgen deficiency in men with HH is treated with testosterone whenever fertility is not currently desired. In teenagers with congenital HH, androgen treatment stimulates body and facial hair growth, penile enlargement, muscle development, voice deepening, libido and potency, morning erections, and nocturnal emissions, and increases the hematocrit. Androgen treatment stimulates GH production by increasing the amplitude of spontaneous secretory episodes19 and thereby contributes to the adolescent growth spurt and increase in bone mass.95 Such physical changes also have an important psychological impact. Only native testosterone preparations are recommended as they provide full virilization, are nonhepatotoxic, and are aromatizable to estradiol (as required for bone health).
Initiation of treatment in adolescents can begin with lower doses to reduce dose-dependent side effects such as acne and gynecomastia; for example, testosterone cypionate or enanthate can be started at 75 mg monthly and gradually increased to a full replacement dose of 200 mg every 2 weeks over a period of 2 to 3 years. Transdermal testosterone gels are an alternative, with initial treatment delivering 2.5 mg daily at bedtime to simulate a normal pubertal nocturnal rise and a gradual increase to full replacement doses. Historically, when the differential diagnosis is between IHH and constitutional delay of puberty, androgen treatment often was postponed until 18 years of age, but given the decreased peak bone mass that occurs with sex steroid deficiency during the teenage years96 and the social ridicule to which androgen-deficient teenagers are exposed, androgen treatment by 14 to 15 years of age is indicated with intermittent withdrawal of therapy sometime thereafter for reevaluation of endogenous androgen production.
In HH, hCG can be used as an LH substitute to stimulate testicular testosterone production with effective virilization, and in contrast to treatment with testosterone, it stimulates testicular growth. Generally, doses of 1000 to 1500 IU given subcutaneously twice weekly sustain adult serum testosterone levels. Induction of testicular aromatase during hCG treatment may produce high serum estradiol levels and gynecomastia. Because of cost factors and the need for frequent injections, hCG treatment usually is initiated only when fertility is desired. Treatment with testosterone for many years does not preclude a favorable response to hCG.97
Although LH and FSH both are required for quantitatively normal spermatogenesis, selected patients with IHH may produce spermatozoa and successfully impregnate their partners when treated with hCG alone. Pretreatment testis size greater than 3 mL indicates some endogenous gonadotropin secretion and may indicate responsiveness to hCG alone.98
Treatment efficacy is assessed by clinical observation and sequential determination of serum testosterone and semen analysis (Fig. 139-3). Among responders, testes grow to a volume of 12 to 15 mL and spermatozoa usually appear in the ejaculate within 12 months of the start of treatment. hCG alone generally is successful in restoring spermatogenesis in men with pituitary or suprasellar tumors that develop postpubertally99 (Fig. 139-4) and also may restore sperm production in men with IHH previously treated successfully with both hCG and FSH.100
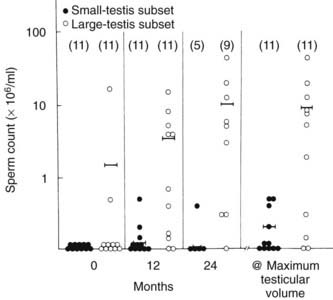
FIGURE 139-3. Mean sperm concentration during human chorionic gonadotropin therapy in 22 men with isolated hypogonadotropic hypogonadism (IHH). Men in the small-testis subset (n = 11) had a mean testis volume of 3 mL or less at the start of the study, consistent with complete IHH.
(Data from Burris AS, Robard HW, Winters SJ, et al: Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size, J Clin Endocrinol Metab 66:1144–1151, 1988.)
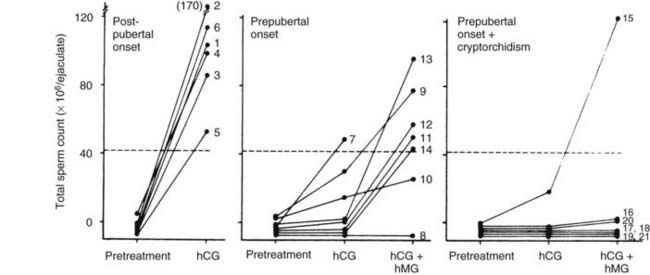
FIGURE 139-4. Effects of treatment with human chorionic gonadotropin (hCG) alone and in combination with human menopausal gonadotropin (hMG) on sperm output in men with gonadotropin deficiency (GD). Men with GD of postpubertal onset had pituitary adenoma (two), craniopharyngioma (three), or unknown cause (one). Cryptorchidism (unilateral in six of seven) had been treated during childhood in men with GD of prepubertal onset.
(From Finkel DM, Phillips JL, Snyder PJ: Stimulation of spermatogenesis by gonadotropins in men with hypogonadotropic hypogonadism, N Engl J Med 313:651–655, 1985.
Most patients with IHH require treatment with FSH as well as hCG to induce spermatogenesis, including patients with pretreatment testis volumes of less than 4 mL and men with larger testes at baseline whose testes fail to grow to volumes of 12 to 15 mL with hCG treatment and who remain azoospermic.98 All men generally are pretreated with hCG for 6 months before FSH is added. Recombinant human FSH now is used widely in favor of urinary derived products at a dose of 100 to 150 IU subcutaneously given three times weekly; more than two thirds of men will become sperm positive (>1 million/mL) if treatment is continued for 18 months,101 with a pregnancy rate of up to 90%.102 Most pregnancies occur with sperm counts well below the normal range of 20 million/mL.103
The absence of Sertoli FSH-induced Sertoli cell division in these men may limit the increase in testis size and result in permanently impaired Sertoli cell maturation, thereby limiting the testicular response to treatment in adulthood. The occurrence of cryptorchidism may diminish the response to gonadotropin therapy, although unilateral cryptorchidism does not preclude fertility104 (see Fig. 139-4).
GnRH also can be used to stimulate spermatogenesis in men with GnRH deficiency.105 Programmable portable infusion pumps delivering pulses of GnRH subcutaneously every 2 hours in line with normal GnRH pulse frequency can restore serum gonadotropin and testosterone levels as well as spermatogenesis in men with GnRH deficiency.105 Sperm appear in the ejaculate in approximately two thirds of patients after 18 to 139 weeks of treatment. One prospective but nonrandomized study compared hCG/FSH treatment versus pulsatile GnRH treatment106 and noted that testis size was increased more by GnRH than by hCG/FSH in men with complete IHH, and sperm appeared earlier (12 versus 20 months, respectively) but final sperm concentrations were similar. A more recent report has confirmed this result.104 Poor patient compliance can be a limitation of pulsatile GnRH therapy, and allergic reactions107 and GnRH-binding antibodies108 may develop. Given the cost, complexity, and inconvenience of pulsatile GnRH therapy, hCG/FSH continues to be recommended as initial therapy, and GnRH is reserved for treatment failures and generally is available only through highly specialized centers.
For those men who achieve a very low sperm count even after prolonged gonadotropin or GnRH therapy, assisted reproduction (specifically, intracytoplasmic sperm injection) provides another avenue for conception. Sperm cryopreservation should be offered after successful induction of spermatogenesis to permit subsequent pregnancies through insemination. If the second pregnancy is planned for shortly thereafter, continuation on hCG alone probably will maintain fertility. Otherwise, gonadotropin treatment should be continued until the beginning of the second trimester to ensure an ongoing pregnancy before reversion back to testosterone replacement therapy.
Primary Testicular Failure
Testicular damage can result from congenital or acquired causes and may be reflected by impairment in spermatogenesis and/or hypoandrogenism; the predominant clinical manifestation varies with the cause (Table 139-5). The testes usually are reduced in size, men are subfertile or sterile, and symptomatic androgen deficiency is relatively common. When testosterone deficiency develops rapidly, as with orchitis, vasomotor symptoms comparable with those of the female climacteric may occur.109 Elevated FSH and LH levels are markers of primary testicular failure, reflecting damage to the seminiferous tubule and Leydig cell components, respectively. Because the seminiferous tubules are more sensitive to damage than Leydig cells, FSH levels often are increased selectively, although a subtle disturbance in LH secretion is suggested by an exaggerated LH response to stimulation with GnRH.110
Table 139-5. Causes of Primary Testicular Failure
| Congenital | Acquired |
|---|---|
| Klinefelter’s syndrome | Trauma |
| 46,XX male | Torsion |
| Other chromosomal aneuploidies | Orchidectomy |
| Y-chromosome deletions | Orchitis |
| Noonan’s syndrome | Chemo/radiotherapy |
| Congenital anorchia | HIV infection |
| Cryptorchidism | Chronic liver disease |
| Myotonic dystrophy | Autoimmune polyglandular failure |
| Hemoglobinopathies | Spinal cord injury |
Gonadotropin production increases because negative feedback inhibition by sex steroids and inhibin B is decreased. The frequency and amplitude of LH secretory episodes are increased in men with testicular failure111 (Fig. 139-5) and are reduced by testosterone replacement.112 Direct sampling of GnRH in hypothalamic portal blood in orchiectomized rams reveals that GnRH pulse frequency is regulated by testosterone. The rise in LH pulse frequency that occurs during treatment with aromatase inhibitors or estrogen receptor antagonists implies that estrogens regulate the GnRH pulse generator in men as well.113
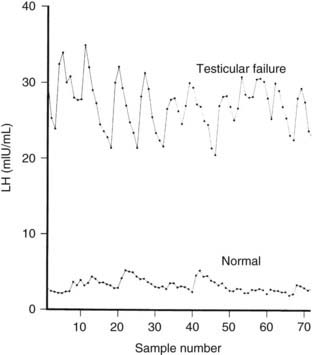
FIGURE 139-5. Luteinizing hormone (LH) levels in serum samples drawn every 10 minutes for 12 hours beginning at 8:00 am in a normal adult man aged 21 years and a 35-year-old man with bilateral cryptorchidism.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


