FIGURE 99-1. Interacting feedback loops controlling aldosterone secretion. Volume is regulated through the renin-angiotensin system, and potassium is regulated through direct feedback. A-I, Angiotensin I; A-II, angiotensin II; BP, blood pressure.
Volume status is sensed by the renin-secreting juxtaglomerular cells of the kidney. Where the sodium status (and hence, volume) is low, renin will be secreted. Renin, an aspartyl protease, is synthesised as inactive prorenin that is activated by the action of a protease. Renin release from the juxtaglomerular cells is influenced by a number of factors (Table 99-1), including renal perfusion pressure, the sympathetic nervous system, and prostaglandins (which are stimulatory), dopamine, atrial natriuretic peptide (ANP), and angiotensin II, all of which are inhibitory. Renin acts on angiotensinogen to release the decapeptide angiotensin I, which in turn is subject to further proteolysis by angiotensin-converting enzyme, primarily in the pulmonary vascular bed, to yield the octapeptide angiotensin II. Angiotensin II acts via its specific G protein–coupled receptor in the vasculature as a potent vasoconstrictor (thereby defending plasma volume and blood pressure) and on the adrenocorticoid glomerulosa cells to stimulate aldosterone synthesis.6 The latter response promotes sodium retention with a consequent increase in plasma volume. Aldosterone biosynthesis in the zona glomerulosa of the adrenal cortex is regulated by transcription of the aldosterone synthase gene (CYP11B2). As with other steroidogenic enzymes, steroidogenic factor-1 (SF-1) is required for aldosterone synthase expression. Members of the NR4A family of nuclear receptors have been shown to be regulators of aldosterone synthase gene expression.7 Although angiotensin II is important in the regulation of aldosterone, a response to low-salt or high-potassium diet is also seen in mice in which the angiotensinogen gene has been deleted.8 In these mice, the regulation of aldosterone is directed primarily by serum potassium levels.
Table 99-1. Factor Regulating Renin Release
| Stimulatory | Inhibitory |
|---|---|
| Decreased perfusion pressure | Increased chloride delivery at the macula densa |
| PGl2 | Angiotensin II |
| ACTH | Atrial natriuretic factor |
| Vasopressin | |
| β-Adrenergic stimulation | α-Adrenergic stimulation |
| Dopamine |
ACTH, Adrenocorticotropic hormone; PGl2, prostacyclin.
The secretion of aldosterone is also stimulated by potassium, so a negative feedback loop exists for potassium and aldosterone. It should be noted that aldosterone also affects acid-base balance by increasing the exchange of hydrogen ions for sodium. Therefore, the net effect of an increase in aldosterone levels, as may result from an aldosterone-producing tumor (Conn’s syndrome) or exogenous mineralocorticoid administration (e.g., 9α-fludrocortisol), is sodium resorption with consequent volume expansion, hypertension and suppression of plasma renin activity, hypokalemia, and a metabolic alkalosis.
Aldosterone secretion is also subject to negative regulation. ANP is a potent inhibitor of aldosterone secretion, consistent with its role to promote natriuresis. Dopamine is a well-characterized inhibitor of aldosterone secretion.9 Other inhibitors have been described, but their physiologic relevance is not clear (Table 99-2).
Table 99-2. Factors Regulating Aldosterone Secretion
| Factor | Stimulatory | Inhibitory |
|---|---|---|
| Peptides | Angiotensin II | Atrial natriuretic peptide |
| Angiotensin III | Somatostatin | |
| ACTH | ||
| Vasopressin | ||
| Endothelin | ||
| Ions | Plasma potassium | |
| Other | Serotonin | Dopamine |
| Ouabain |
ACTH, Adrenocorticotropic hormone.
Dietary sodium has a major impact on the state of the renin-angiotensin-aldosterone system (RAS). Sodium deficiency increases adrenal sensitivity to angiotensin II over time; the converse is true of the vasopressor response. Aldosterone-induced sodium retention restores volume status by maintaining the balance between volume and capacity.10
The response of the individual to aldosterone-mediated sodium retention is self-limiting in that after 3 to 4 days, expansion of the extracellular volume plateaus and of sodium secretion returns to control levels. This process is termed escape.11 It should be noted that the kaliuretic effect persists despite the escape of sodium retention. Intrarenal regulators, particularly prostaglandins, are probably the critical mediators of the escape, although other factors (e.g., ANP) may play a role.12,13
A local renin-angiotensin system has been reported to operate in a number of tissues, including the submaxillary glands, gonads, smooth muscle cells, adipose tissue, pituitary, brain, and adrenal cortex. The existence of this system is often determined by the presence of mRNA for renin, angiotensinogen, and angiotensin-converting enzyme (ACE); the relative physiologic importance of these local systems has recently been called into question.14
Potassium Homeostasis
Aldosterone is primarily involved in the chronic regulation of plasma potassium levels.15 Acute regulation involves nonrenal mechanisms such as those mediated by insulin and β-adrenergic agonists. Aldosterone regulates potassium homeostasis through direct effects on transport of epithelia, including its effects on sodium homeostasis. Small fluctuations in plasma potassium influence aldosterone secretion. Although the mechanism of these effects has not been determined, it is known to be independent of angiotensin II levels; however, plasma potassium levels do alter the sensitivity of the adrenal to angiotensin II. The local adrenal RAS has been implicated in the adrenal response to potassium; the circulating system is inhibited by potassium, whereas local adrenal production is increased.
Aldosterone secretion is also subject to regulation by adrenocorticotropic hormone (ACTH); however, aldosterone regulation is normal in patients with hypopituitarism.16
Mineralocorticoid Receptors
The classic actions of aldosterone involve epithelial cells in the distal nephron and distal colon; these mediate sodium flux. As with other steroid hormones, the principal mode of action, at least in sodium transport, involves an intracellular receptor that when activated by ligand-binding regulates gene transcription, a so-called genomic mechanism of action.
High-affinity cytosol and nuclear binding of 3H-aldosterone were first described in classic mineralocorticoid target tissues such as kidney5,17 and parotid18 more than 30 years ago. Spironolactone was shown to block aldosterone binding and action on urinary electrolytes in parallel,19 providing evidence that these sites are physiologic mineralocorticoid receptors (MRs). MR were subsequently cloned from human kidney,20 and the rat homologue was cloned from a hippocampal cDNA library.21 In contrast to glucocorticoid receptors (GRs), which are expressed ubiquitously, MRs have a tissue-specific pattern of expression, with highest levels observed in the distal nephron,22 distal colon,23 and hippocampus.20 Lower levels of expression are observed elsewhere in the gastrointestinal tract; in cardiovascular tissues; and in a range of other tissues, both epithelial and nonepithelial.20,22,23
The human mineralocorticoid receptor is a protein of 984 amino acids and, together with the GR, progesterone receptor (PR), and androgen receptor (AR), forms a distinct subfamily within the steroid/thyroid/retinoid/orphan receptor superfamily.24 This receptor superfamily is defined by a central cysteine-rich DNA-binding domain. At the C terminus is the ligand-binding domain (LBD), which has a highly conserved tertiary structure. The N-terminal domain has little or no homology between receptors. Within the MR/GR/PR/AR subfamily, MR and GR are closely related, with 94% amino acid identity in the central cysteine-rich DNA-binding domain, and 57% identity in the C-terminal ligand-binding domain (Fig. 99-2). The MR and GR, however, are located on different chromosomes (MR on 4q31.225; GR on 5q3126).
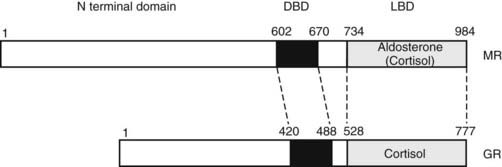
FIGURE 99-2. Domain structure of the mineralocorticoid receptor (MR) and the closely related glucocorticoid receptor (GR) showing the three principal functional domains: the N-terminal domain, the DNA-binding domain (DBD), and the ligand-binding domain (LBD). These domains share less than 15%, 94%, and 57% identity, respectively.
The cysteine residues of the DNA-binding domain complex around two zinc atoms to form two α-helices, one of which lies in the major groove and binds with a common consensus sequence in the DNA, the hormone response element.27 The LBD consists of 11 α-helices, which form a three-layered structure with the ligand-binding pocket buried in the middle.28–30 The N-terminal domain contains a transcription activation function that is relatively unstructured. The N-terminal domains are not conserved between steroid receptors, although in several (including the MR31), a functional interaction has been described between the N-terminal and ligand-binding domains. It is curious that for the MR, this interaction is seen only with aldosterone; cortisol acts as an antagonist.
The unliganded receptor is predominantly cytoplasmic,32 being complexed with the heat shock proteins 70 and 90 and their co-chaperones.33 This configuration maintains the receptor in a transcriptionally inactive high-affinity binding state. The interaction of the ligand-binding domain with this complex is an important determinant of ligand-binding affinity and specificity.34 The mineralocorticoid receptor antagonists, spironolactone and eplerenone, appear to be accommodated into the ligand-binding pocket without distortion,35 suggesting that the mechanism of their antagonism differs from that of the estrogen receptor antagonists such as tamoxifen and raloxifene. These latter compounds exhibit tissue-specific antagonism, in contrast to spironolactone, which is a pure antagonist. Evidence that cortisol/corticosterone may antagonize the actions of aldosterone at the MR in certain tissues suggests that different ligands may induce differing conformations on binding the receptor.31 At a cellular level, these differing conformations result in differential interactions with the transcriptional machinery through the mediators of this signaling, the co-regulatory molecules. In contrast to the other steroid receptors, such interactions are only now being characterized for the MR.36
Very good evidence suggests that not all MRs are physiologic receptors for aldosterone. In brief, cortisol and corticosterone (the physiologic glucocorticoid in the rat) have an affinity for the MR equivalent to that for aldosterone and substantially higher than their affinity for the GR.20 MRs are distributed widely in tissues in which a physiologic effect of aldosterone on Na+ homeostasis is unlikely (e.g., the hippocampus20,37). Given the much higher circulating levels of glucocorticoids than of aldosterone, these sites appear to be high-affinity GRs in such tissues. In nonepithelial tissues, the response of MR to cortisol/corticosterone and aldosterone often is not equivalent,38 leading to speculation about the relationship of epithelial to nonepithelial MR. Most evidence to date would suggest that although the MR gene uses multiple, tissue-specific promoters,39–41 the coding region is unaltered between tissues, with the possible exception of some minor isoforms.42 The explanation for such differences between tissues may lie in the nature of the conformation that the MR adopts after ligand binding.43 Such conformational differences may alter some but not all transactivation functions, such that tissue-specific receptor co-activators or co-repressors36 may mediate different responses in different tissues.
A second question, given the equivalent affinity of MR for glucocorticoids and aldosterone, is that of the mechanism(s) allowing aldosterone occupancy of MR in physiologic mineralocorticoid target tissues. This matter is discussed later in this chapter. Not only do MRs appear unable to distinguish between physiologic mineralocorticoids and glucocorticoids, but evidence suggests an equivalent lack of selectivity at the level of the response elements, where both MR and GR act as transcription factors.20,44 Because no MR-selective response elements have been characterized to date, the evidence for this lack of specificity is indirect but clearly established for epithelial tissues.45 In vitro studies on cultured cortical collecting tubule cells have shown aldosterone, dexamethasone, and the highly specific GR agonist RU28362 to have indistinguishable effects on unidirectional Na+ and K+ fluxes and on the transepithelial potential difference as measured by short-circuit current.46 In vivo studies on adrenalectomized rats given the highly specific GR agonist RU28362 similarly have shown that GR, appropriately activated by ligand, can activate the same genes as the MR2 and can produce a classic mineralocorticoid effect on urinary electrolytes. On the other hand, differences in the action of MR and GR in the same cells can be demonstrated,44,45,47,48 suggesting the possibility of greater complexity in certain circumstances (e.g., when GR but not MR can be shown to interact with other transcription factors such as adaptor protein-1 [AP-1]).47
In addition to its effects on ion flux in classic mineralocorticoid target tissues, aldosterone has been shown to have effects via MR occupancy in a variety of other tissues. Aldosterone elevates blood pressure in the rat when infused into the cerebral ventricles49; this effect clearly results from unprotected MR, because it is blocked by simultaneous infusion of low doses of corticosterone. Thus, corticosterone in the AV3V region acts as an aldosterone antagonist on MR, in contrast with the kidney and other epithelia, where its action is to mimic aldosterone.46,50 An additional difference between epithelial and nonepithelial tissues is that in the former, activation of GR has been shown to mimic that of MR,46,50 whereas in nonepithelial tissues, this clearly is not the case. In the heart, a nonepithelial tissue that expresses MR, studies conducted in vivo show that levels of aldosterone inappropriately high for the Na+ status of the rat produce diffuse perivascular and interstitial fibrosis, an effect that can be antagonized by corticosterone or spironolactone.51 The clinical correlation of these observations is found in two recent large trials, which show a benefit of the addition of a mineralocorticoid antagonist to the conventional regimen in the treatment of individuals with severe cardiac failure, with respect to both morbidity and mortality.52,53
Mice homozygous for inactivating mutations in the MR gene54 (MR knockout, or MRKO) show classical features of aldosterone deficiency—salt-wasting, hyperkalemia, and dehydration—but have marked hyperaldosteronism; these features are also seen in the syndrome of pseudohypoaldosteronism (PHA) (see Chapter 108). MRKO mice are born at the expected frequency from heterozygote matings; untreated, they begin to deteriorate from day 5, and they die between day 8 and day 11; treatment by salt supplementation allows survival and normal growth. Mutations of the MR have been reported in the autosomal dominant form of PHA,55–57 which thus appear to be equivalent to mice heterozygous for the MR gene knockout. In the more severe autosomal recessive form and in many sporadic cases of PHA, mutations of the MR are not observed.56,57
GENOMIC VERSUS NONGENOMIC ALDOSTERONE ACTIONS
Considerable interest has been expressed with respect to steroid hormone action in terms of whether all responses are mediated through the classical nuclear receptor with direct regulation of gene expression, or whether other pathways, perhaps involving novel cell membrane receptors, exist58; the evidence for novel receptors is not compelling.59 Clear evidence has been found both in vitro and in vivo for rapid nongenomic signaling. This can involve activation by src kinase of the epidermal growth factor (EGF) receptor with consequent downstream signaling through the mitogen-activated protein (MAP)-kinase pathway; the signaling appears to require only the LBD of the MR.60 McEneaney et al.61 defined rapid effects on protein kinase signaling, Mihailidou et al.62 reported rapid effects in isolated cell patches from cardiomyocytes, and Alzamora et al.63 observed rapid effects in vascular cells. Karst et al.64 found rapid nongenomic effects of corticosterone on glutamate release from the CA1 pyramidal neurons of the hippocampus. This response also involves mitogen-activated protein kinase/extracellular signal–related kinase (MAPK/ERK) signaling.65 In each case, the receptor involved is the classical MR. The relative contribution to the mineralocorticoid response by this signaling has not yet been evaluated, although it is speculated that this rapid response may prime the transcriptional response65 or may alter the dynamic range of the response.65
Specificity-Conferring Enzymes
11β-HYDROXYSTEROID DEHYDROGENASE TYPE 2
Although the affinity of MR in vitro20,37 is equal for aldosterone, corticosterone, and cortisol, in vivo cortisol is excluded from such receptors in the kidney, parotid, and colon (but not in the hippocampus).66 This reflects the activity of the enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD), which is responsible for the interconversion of cortisol and cortisone (in the rat, corticosterone and 11-dehydrocorticosterone). In the kidney, the predominant direction of conversion is cortisol to cortisone, as is shown by the reduced cortisol-to-cortisone ratio in human renal venous blood.67 Initially, rat liver 11β-HSD was purified, cloned, and sequenced,68 as was its human homologue69; it is expressed at high levels in the liver, testis, lung, and renal proximal tubule,70 none of which are physiologic mineralocorticoid target tissues. Subsequently, a second isoform (11β-HSD2, with the “liver” isoform termed 11β-HSD1) was isolated by expression cloning71 and was shown to be the enzyme responsible for the aldosterone selectivity of MR in epithelial aldosterone target tissues.72,73 Unlike 11β-HSD1, it co-localizes with MR32 in renal distal tubular elements, colon, sweat, and salivary glands74; it is also expressed at high abundance in the placenta71 and in select areas (subcommissural organ, ventromedial ventrolateral hypothalamus) of the rat brain.75 It has a low Km (i.e., high affinity) for both corticosterone (≈5 nM) and cortisol (≈50 nM), unlike the micromolar Km of 11β-HSD1; in vivo, it appears operationally unidirectional, acting uniquely as a dehydrogenase, whereas 11β-HSD1 appears to act predominantly as a reductase.72 Deletion of the 11β-HSD2 gene in mice yields a phenotype76 consistent with the clinical syndrome of human apparent mineralocorticoid excess (AME).72,73,77
In AME, 11β-HSD2 activity is congenitally low, indicated by a markedly increased ratio of urinary cortisol to cortisone metabolites; abnormally high intrarenal cortisol levels occupy receptors normally protected by the enzyme, resulting in increased Na+ retention and elevation of blood pressure.72,73,78 Kindred with AME have been examined for the presence of mutations in the gene coding for 11β-HSD2.77,79,80 The mutations are autosomal recessive and thus are commonly seen in the context of consanguinuity, although compound heterozygosity has also been reported.77 Although heterozygotes may show subtle departures from normal in terms of their ratio of urinary cortisol to cortisone metabolites, little evidence indicates a substantial deficit in such individuals (see also Chapter 108).
Licorice ingestion has long been known to cause Na+ retention, hypokalemia, and hypertension; the mechanism of its action was elucidated by elegant studies78 on human volunteers, in whom ingestion of 250 g of licorice a day for 10 days produced a clinical picture equivalent to a mild form of apparent mineralocorticoid excess. When rats were given glycyrrhetinic acid (the active principal of licorice)81 or carbenoxolone (glycyrrhetinic acid hemisuccinate)82 to block 11β-HSD, the normal aldosterone selectivity of epithelial MR was abolished.
These studies demonstrate the crucial specificity-conferring role in mineralocorticoid target tissues for 11β-HSD2. Aldosterone is not a substrate for 11β-HSD, because its 11β-OH group is stabilized as an 11,18-hemiketal via cyclization with the highly reactive aldehyde group unique to aldosterone at C18.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


