FIGURE 14-1. Synthesis and secretion of growth hormone by the anterior pituitary gland.
(Modified from Melmed S: Acromegaly. New Engl J Med, 355:2558–2573, 2006.)
EXCESS GROWTH HORMONE SECRETION
Tumors may arise from clonal expansion of one or more of the anterior pituitary differentiated cell types22,23 and thereby result in specific hormone hypersecretory syndromes. The most common cause of acromegaly is a somatotroph (GH-secreting) adenoma of the anterior pituitary, which accounts for 30% of all hormone-secreting pituitary adenomas1 (Fig. 14-2). GH-secreting adenomas arise from differentiated cells secreting GH gene products1,23,24 (Table 14-1). These cells include somatotrophs, mixed mammosomatotrophs (secreting both GH and prolactin [PRL]), or more primitive acidophilic stem cells. Regardless of their cellular origin, transformation and subsequent replication of these cells result in adenoma formation as well as unrestrained GH secretion.1 Most patients harbor densely granulated GH-cell adenomas, which are commonly encountered in older patients with indolent disease progression. Sparsely granulated GH-cell adenomas occur in younger patients with more aggressive disease onset and higher GH levels.1,2 Mammosomatotroph cell tumors, or discrete mammotroph and somatotroph tumors, reflect the common stem cell origin of the somatotroph cell lineage.2,24–26 Although acidophil stem cell adenomas secrete GH, their predominant product is PRL, thus accounting for the high incidence of hyperprolactinemic symptoms (galactorrhea, amenorrhea, infertility) initially seen in these patients.27 Patients with McCune-Albright syndrome also may have acromegaly, although the presence of a discrete GH-cell adenoma has been inconsistently reported in these cases.28 Rarely, acromegaly may occur in patients with a partially empty sella.29 The rim of pituitary tissue surrounding the empty sella may harbor a small endocrinologically active GH-secreting adenoma not visible on magnetic resonance imaging (MRI) (i.e., <2 mm in diameter). Because embryonic pituitary tissue originates from the nasopharyngeal Rathke’s pouch, ectopic pituitary adenomas may arise in remnant nasopharyngeal tissue along the line of primitive adenohypophysial migration. These adenomas may not be detected on pituitary MRI fields, and more extensive skull base imaging may be required. Very rarely, ectopic GH production by pancreatic,30 lung,31 ovarian,32 or lymphocytic neoplasms may result in acromegaly.1
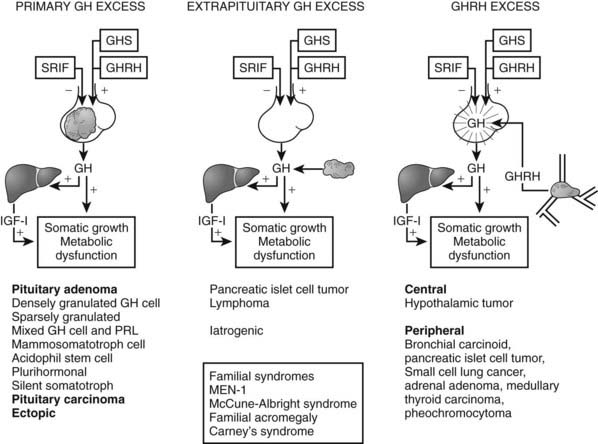
FIGURE 14-2. Pathogenesis of acromegaly.
(Modified from Melmed S: Acromegaly, New Engl J Med 355:2558–2573, 2006.)
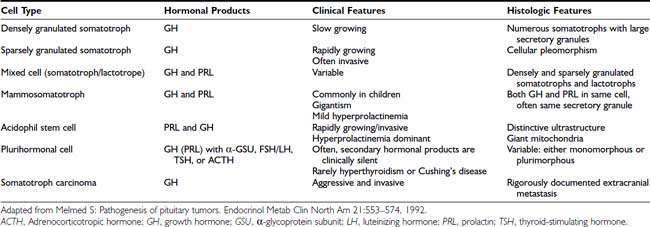
EXCESS GROWTH HORMONE–RELEASING HORMONE SECRETION
Excessive circulating levels of GHRH may overstimulate the pituitary and cause somatotroph hyperplasia, GH hypersecretion, and acromegaly.33 Central overproduction of GHRH may occur in patients harboring hypothalamic hamartomas or gangliocytomas.33 These rare tumors are usually diagnosed by pathologic examination of a surgically resected sellar mass causing GH hypersecretion and acromegaly.34 Ectopic GHRH production by carcinoid tumors, although rare, accounts for most cases of acromegaly.35 The clinical association of acromegaly with carcinoid disease had long been recognized, and the pathogenesis of ectopic GHRH production is now elucidated. Subclinical GHRH immunoreactivity has been demonstrated in about 40% of lung, abdominal, and bony carcinoid tissue specimens.
Pituitary somatotroph hyperplasia plus acromegaly associated with ectopic GHRH production has been reported in more than 100 patients, and the original isolation of GHRH was accomplished from a pancreatic carcinoid tumor.36 Because the peripheral features of hypersomatotropism are quite similar in all forms of pituitary and nonpituitary acromegaly, diagnosis of the etiology of the disease may be clinically challenging.1
Acromegaloidism is a very rare syndrome characterized by acromegalic features with no discernible pituitary tumor and normal serum GH and IGF-1 concentrations. It has been presumed that this disorder is due to excess secretion of a putative, as yet unidentified, growth factor.37
ROLE OF THE HYPOTHALAMUS IN THE ETIOLOGY OF ACROMEGALY
Hypothalamic GHRH and SRIF selectively regulate GH gene expression and secretion.8 These hypothalamic peptide hormones are expressed both within the anterior pituitary gland itself and within GH-secreting pituitary tumors.38,39 GHRH, in addition to its hormonal regulation of GH production, induces somatotroph DNA synthesis.40 Mice bearing an overexpressing GHRH transgene are subject to somatotroph hyperplasia and ultimately to pituitary adenomas.41,42 In patients with carcinoid tumors and ectopic GHRH production, somatotroph hyperplasia and occasionally adenomas also may develop, which suggests that disordered endocrine or paracrine GHRH or SRIF action may be permissive for pituitary tumor growth.43 GHRH signaling defects also have been identified in acromegaly. Constitutive activation of the GHRH receptor G-protein signaling unit facilitates ligand-independent induction of GH gene expression. This gsp mutation results in guanosine triphosphatase (GTPase) inactivation, with subsequent elevated cyclic adenosine monophosphate (cAMP) levels and GH hypersecretion.44,45 Excessive CREB (cAMP response element binding protein) serine phosphorylation also may account for activation of the CREB–Pit-1 (pituitary-specific transcription factor 1) signaling unit in a subset of GH-cell adenomas.46
Pituitary tumor–derived paracrine GHRH or SRIF or both also may regulate tumor growth or function, although constitutively activating hormone receptor structural mutations have not been identified clinically. A truncated alternatively spliced GHRH receptor transcript has been described, but its functional significance is unclear.47 In light of compelling evidence favoring intrinsic genetic defects occurring in GH-secreting pituitary tumors, as discussed later, it is apparent that hypothalamic influences may be permissive of tumor growth rather than being proximally involved in the initiation of somatotroph tumorigenesis.43,48
INTRINSIC PITUITARY LESIONS
Virtually all GH-cell adenomas arise as discrete clonal expansions of a transformed cell22 (Table 14-2). This monoclonal origin implies that intrinsic genetic alterations account for tumorigenic initiating events and supports abundant earlier clinical observations that resection of small well-circumscribed adenomas usually results in surgical cure of GH-secreting adenomas.24,43,49 Because adenohypophyseal tissue surrounding the pituitary adenoma is histologically normal, it is unlikely that multiple independent cellular growth events (e.g., generalized hyperplasia) precede adenoma formation. Increasing evidence points to complex molecular cascades accounting for the cellular progression, resulting in pituitary-cell transformation and, ultimately, tumor formation. Multistep development of pituitary acromegaly involves a spectrum of genetic alterations associated with dysregulation of cell proliferation, differentiation, and GH production.43 Activation of oncogene function or inactivation of tumor-suppressor genes or both may account for these changes43,50,51 (see Table 14-2).
Table 14-2. Evidence for an Intrinsic Pituitary Defect in the Pathogenesis of Acromegaly
GH, Growth hormone.
Adapted from Drange MR, Melmed S: IGFs in the evaluation of acromegaly. In Rosenfeld RG, Roberts CT (eds): Contemporary endocrinology. The IGF system: molecular biology, physiology, and clinical applications. Totowa, NJ, 1999, Humana, pp 699–720.
CANDIDATE GENES IN THE ETIOLOGY OF ACROMEGALY
Inactivating Mutations
Several transgenic animal models have shown that disruption of tumor-suppressor genes (including RB [retinoblastoma] and p27) results in a high incidence of pituitary tumor formation in afflicted mice.52–54 Because a variety of chromosomal loss of heterogeneity (LOH) patterns are observed in human adenomas, loss of tumor-suppressor gene activity was similarly postulated for human tumors (Table 14-3).
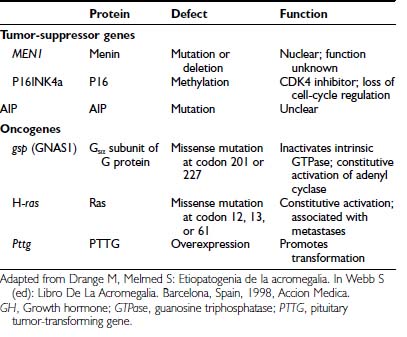
Several chromosomal lesions occur in pituitary tumor tissue derived from patients with sporadic nonfamilial acromegaly. LOH involving chromosomes 11q13, 13, and 9 occurs in up to 20% of sporadic50,55,56 pituitary tumors. Despite the multiple endocrine neoplasia type 1 (MEN1) gene location on chromosome 11, non-MEN1 patients with sporadic pituitary tumors and 11q LOH harbor intact coding and intronic sequences, with appropriately expressed MEN1 messenger RNA (mRNA).56 Lesions in chromosomes 13 and 9 also are more prevalent in invasive or larger adenomas.51 Chromosome 13q LOH occurs in proximity to the RB locus and was found in 13 aggressive pituitary tumors, whereas small circumscribed tumors exhibit intact RB alleles.57 These results suggest the presence of putative tumor-suppressor genes located on chromosomes 11 and 13 that may be involved in controlling the propensity for pituitary tumor proliferation. Despite these heterogeneous chromosomal LOH patterns, consistent loss of tumor-suppressor gene activity has not been identified for acromegaly. Although tumor invasiveness or size correlates with an increased propensity for chromosomal LOH,51 identification of a specific molecular lesion leading to loss of antiproliferative activity in GH-secreting tumors remains elusive43 (see Table 14-2).
Activating Mutations
GTPase acts to inactivate stimulatory G (Gs) proteins that induce adenyl cyclase and intracellular cAMP accumulation.58 Missense mutations replacing residue 201 (Arg → Cys or His) or 227 (Gln → Arg or Leu), termed gsp, result in persistently elevated ligand-independent Gs activity and constitutively elevated cAMP and GH hypersecretion.44 Gsp mutations occur in a subset of GH adenomas, with a prevalence ranging from 30% to 40% in whites58–63 to only 10% in Japanese64 patients with acromegaly. Clinical or biochemical correlations have not been associated with gsp mutations.44,65 Thus, although these mutational events suggest a compelling mechanism for explaining GH-cell hypersecretion, their clinical significance has not been apparent (see Table 14-3), because the natural course of the disease does not differ in gsp+ve or gsp−ve patients.
Rarely, ras mutations have been observed in highly invasive pituitary tumors or their extrapituitary metastases.66–68 Development of true GH-cell carcinoma with documented extracranial metastases, however, is exceedingly rare67 (see Table 14-3). A pituitary tumor-transforming gene (PTTG) was isolated from rat GH-secreting pituitary tumor cells69 and is functionally homologous to yeast securin, which regulates sister chromatid separation during mitosis.70 PTTG overexpression results in cell transformation in vitro and experimental pituitary tumor formation in vivo. PTTG mRNA is abundant in GH-producing tumors, with more than 10-fold increases evident in larger tumors.71 The strong transforming potential of PTTG indicates a role in early induction of GH-cell transformation, possibly by regulating the pituitary cell cycle70 (see Table 14-3).
Familial Syndromes
Acromegaly may occur as a component of MEN syndromes, including the Carney complex or MEN1. The Carney complex consists of myxomas, spotty skin pigmentation, and testicular, adrenal, and pituitary tumors.72–75 About 20% of patients with this autosomal-dominant syndrome associated with chromosome 2p16 harbor GH-secreting pituitary tumors.50,72
The MEN1 gene is located on chromosome 11q13, and LOH of chromosome 11q13 occurs in pancreatic, parathyroid, and pituitary tumors of patients with MEN1.56,76 Inactivation of the MEN1 tumor-suppressor gene likely accounts for the syndrome, in accordance with Knudson’s “two hit” theory whereby both inherited allelic germline mutations and a somatic deletion are required for inactivation of both specific alleles and subsequent tumor formation.77 MEN1, an autosomal-dominant syndrome, consists of hyperplastic or adenomatous parathyroid glands, endocrine pancreas, and anterior pituitary.76 Pituitary adenomas develop in almost half these patients, with GH-cell adenomas reported in about 10% of afflicted subjects.
Isolated familial acromegaly or gigantism not associated with MEN has rarely been reported.50,78,79 Chromosome 11q13 LOH with no discernible MEN 1 mutation was detected in the pituitary adenomas of two brothers with gigantism.80 Low prevalence germline mutations of the aryl hydrocarbon receptor–interacting protein (AIP, located on 11q 13.3) gene were reported as predisposing to a subset of patients with familial acromegaly and gigantism205; 15% of families with isolated familial acromegaly exhibit AIP mutations, and tumors are encountered earlier in subjects harboring a mutation.206,207 AIP has also been proposed as a tumor-suppressor gene for pituitary adenomas.209 The clinical significance of these findings to the broad population of sporadic GH-secreting tumors is at present unclear.208
EPIDEMIOLOGY OF ACROMEGALY
Acromegaly is a rare disease, and accurate assessment of its prevalence in the community has been difficult to ascertain. In Newcastle, England, an annual incidence of 2.8 new patients per million adult population was reported, with an approximate point prevalence of 38 cases per million adult population.81 A higher incidence was reported in Sweden, where the average prevalence of the disease was reported to be 69 cases per million.2,82 If these data are projected to the population of the United States, 750 to 900 new cases would be expected annually, and GH-secreting pituitary adenomas would be present but undiagnosed in another 10,000 to 20,000 persons. The mean age at diagnosis is 40 to 45 years, and its insidious onset may cause the disease to not be diagnosed until 10 to 12 years after symptom onset.82–84 This long delay in diagnosis is often due to the subtle and slow onset of common symptoms, including headache, joint pains, jaw malocclusion, or mild type 2 diabetes. Furthermore, this relatively long time delay allows prolonged exposure of peripheral tissues to unacceptably elevated GH and IGF-1 levels.
Diagnosis
Persistent GH hypersecretion is the hallmark of acromegaly. Excess GH stimulates hepatic production of IGF-1, which is responsible for most of the clinical manifestations of acromegaly.85–87 The diagnosis is often delayed for up to 12 years because of slow clinical progression over many years. Heightened clinical awareness may result in earlier diagnosis.211 Although serum GH and IGF-1 concentrations are both increased in virtually all patients with acromegaly, serum IGF-1 levels may be discordant with GH increases. When a patient is suspected to have acromegaly, biochemical testing is required to confirm the clinical diagnosis, and imaging techniques are used to localize the cause of excess GH secretion (Table 14-4).
Table 14-4. Diagnosis of Acromegaly
GH, Growth hormone; IGF-1, insulin-like growth factor 1; MRI, magnetic resonance imaging.
DOCUMENTING GROWTH-HORMONE HYPERSECRETION
The diagnosis of acromegaly is confirmed by measurement of serum GH after a glucose load and by assessing levels of GH-dependent circulating molecules such as IGF-1 and IGF-binding protein 3 (IGFBP-3).29 IGF-1 levels reflect the integrated bioeffects of GH hypersecretion, and age- and gender-matched elevated IGF-1 levels are pathognomonic of acromegaly.88
Measurement of the serum IGF-1 concentration is the most precise screening test for acromegaly. Unlike those of GH, serum IGF-1 concentrations do not fluctuate hourly according to food intake, exercise, or sleep, but rather reflect integrated GH secretion during the preceding day or longer. Serum IGF-1 concentrations are elevated in virtually all patients with acromegaly, thus providing excellent discrimination from subjects without acromegaly.2,85 In normal subjects, serum IGF-1 concentrations are highest during puberty and decline gradually thereafter; values are significantly lower in adults older than 60 years than in younger subjects. Females have higher levels than do males, and pregnancy may also be associated with elevated IGF-1 levels. Thus, an inappropriately controlled “normal” IGF-1 value in an elderly male patient may in fact be truly elevated and indicative of acromegaly. Serum GH should be measured in patients with equivocal or elevated age- and sex-adjusted serum IGF-1 values.
Although all patients with acromegaly have increased GH secretion, it may be difficult to distinguish elevated random GH levels from normal. As GH levels fluctuate widely throughout the day and night, measuring random GH levels rarely provides useful information for diagnosis of the disorder.2 Short-term fasting, exercise, stress, and sleep are associated with elevated GH, and the availability of ultrasensitive GH assays has indicated that this pulsatile GH rhythm may occur at levels below the detectable sensitivity of previously available assays. Serum GH concentrations fluctuate widely, from less than 0.5 ng/mL (with ultrasensitive assays) during most of the day to as high as 20 or 30 ng/mL at night or after vigorous exercise. Random serum GH concentrations may be elevated in patients with uncontrolled diabetes mellitus, liver disease, and malnutrition, so dynamic tests have been proposed to confirm pituitary GH hypersecretion. The mean GH concentration obtained from 6-hourly samplings will generally provide an integrated summation of net GH secretion, and averaged pooled levels greater than 5 ng/mL are usually encountered in acromegaly.2
The diagnostic hallmark of excess GH hypersecretion was failure to suppress GH levels to 0.4 ng/mL or less (using a chemiluminescent immunoradiometric assay) during a 2-hour period after a 75-g oral glucose load.29 Several factors determine the measurement of serum GH values, including age, gender, BMI, and the type of assay employed. Spontaneous GH secretion is attenuated by 50% to 70% in subjects aged 65 years or more, and IGF-1 levels also decline progressively with age.200 GH levels also correlate inversely with BMI, and lean subjects exhibit higher GH values, as do female subjects.201 Accordingly, criteria for the diagnosis of acromegaly requires demonstrating a mean 24-hour GH level of >2.5 µg/L, a nadir GH of >1 ng/mL after a glucose load, and/or an elevated age- and gender-watched serum IGF-1 level.202
When GH levels were measured by different assays in 46 patients with controlled18 or uncontrolled28 disease,203 values obtained with Diagnostic Products Corporation’s IMMULITE assay were ∼2.3 fold higher than those determined by the Nichols assay. Using the IMMULITE assay, postglucose values of <1 µg/L were associated with disease control, while with the Nichols assay, the proposed cutoff value was 0.5 µg/L. Thus, interpretation of biochemical control should also be determined by knowledge of the specific GH assay employed, as well as the appropriateness of assay controls.204
Invariably, patients who fail to suppress GH after glucose exhibit elevated total IGF-1 levels, with a strong log-linear association between the 24-hour mean GH output and IGF-1 levels.2,89 About 10% or fewer patients may have apparently “normal” GH or IGF-1 levels or both at the time of diagnosis. Repeating the assays in a reputable laboratory may often resolve an apparent clinical/biochemical discordance. Alternatively, reinterpretation of a glucose suppression test or use of a rigorous GH assay may confirm the diagnosis.
Because IGFBP-3 secretion is GH dependent, concentrations may be elevated in patients with acromegaly, thus suggesting that IGFBP-3 measurement may prove useful in diagnosis.90 However, in contrast to the tight correlation of integrated mean 24-hour serum GH with total and free IGF-1 levels, IGFBP-3 levels do not correlate as tightly with disease activity.91,92 Thirty-two percent of subjects with active acromegaly had normal IGFBP-3 levels, and in patients who failed to suppress GH, no consistent elevation of IGFBP-3 was observed.91 Thus, the utility of IGFBP-3 measurements for acromegaly diagnosis or follow-up is limited.
LOCALIZING THE SOURCE OF EXCESS GROWTH HORMONE
Once a biochemical diagnosis of GH hypersecretion is confirmed, MRI of the pituitary to localize the source of hormone excess is indicated. MRI effectively delineates soft-tissue pituitary masses, and gadolinium-enhanced MRI may detect adenomas 2 mm in diameter. In about 75% of patients, the tumor is a macroadenoma (tumor diameter of ≥10 mm) and may extend to parasellar or suprasellar regions or invade the cavernous sinus. More than 90% of patients exhibit a discrete pituitary adenoma on MRI, whereas about 10% of patients may harbor a partial or even apparently total empty sella. Functional GH-secreting adenomas may arise in the remnant rim of pituitary tissue surrounding the empty sella and may not be visible on MRI. Rarely, other nonpituitary causes of acromegaly (see earlier) will require abdominal or chest imaging to localize the source of ectopic GHRH or, more rarely, GH production. Lateral skull radiographs with sellar coned-down tomography or pituitary computed tomographic scans are not usually indicated, because they expose patients to unnecessary ionizing radiation and, when compared with MRI techniques, are insensitive, especially in delineating soft-tissue changes.
Nonpituitary Acromegaly
Rare nonpituitary causes of acromegaly include a hypothalamic tumor secreting GHRH,33,93 a nonendocrine tumor secreting GHRH,36,94 or ectopic GH secretion by a nonendocrine tumor.1,31,32 MRI of the head and pituitary should identify some of these tumors. If pituitary MRI findings are normal, abdominal and chest imaging should be performed, followed by catheterization studies in an attempt to demonstrate an arteriovenous GHRH gradient over the suspected tumor bed. In patients with ectopic GHRH secretion, serum GHRH and GH concentrations are both elevated, and pituitary MRI reveals a normal-sized or enlarged hyperplastic gland.95
An algorithm for the diagnostic evaluation of patients suspected of having acromegaly is shown in Fig. 14-3. A normal age- and gender-controlled serum IGF-1 concentration is strong evidence excluding the diagnosis of acromegaly. If the serum IGF-1 concentration is high (or equivocal), serum GH should be measured within 2 hours after oral glucose administration. If pituitary MRI fails to reveal the presence of a discrete adenoma in the presence of clear-cut biochemical evidence of hypersomatotropism, studies to identify the rarely encountered GHRH- or GH-secreting tumors should be undertaken.
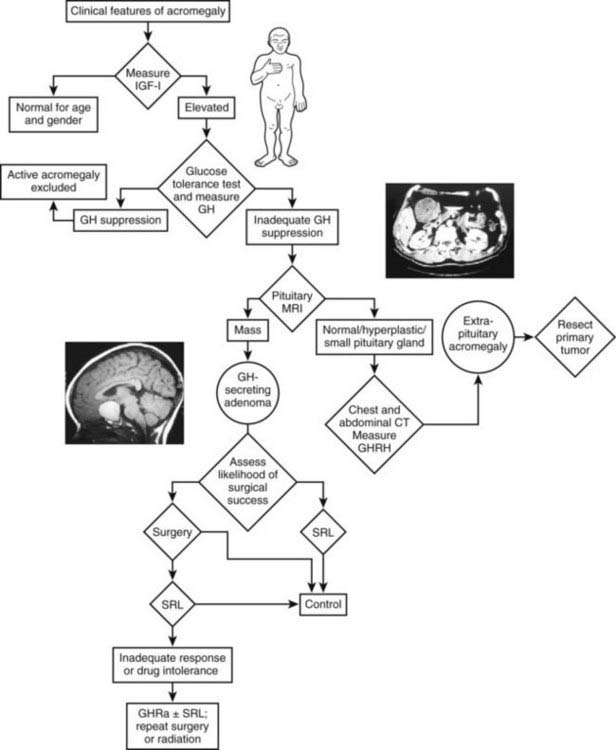
FIGURE 14-3. Diagnosis and management of acromegaly. GH, Growth hormone; GHRH, growth hormone–releasing hormone; IGF-1, insulin-like growth factor 1; MRI, magnetic resonance imaging; OGTT, oral glucose tolerance test; GHRA, GH receptor antagonist; SRL, somatostatin-receptor ligand.
(Modified from Melmed S: Acromegaly, New Engl J Med 355:2558–2573, 2006.)
CLINICAL MANIFESTATIONS
The somatotroph adenoma itself, especially if a macroadenoma, may cause local symptoms such as headache, visual-field defects (classically bitemporal hemianopia), and cranial-nerve palsies. These compressive features are not unique to acromegaly and may occur with any enlarging sellar mass. Nevertheless, the headache associated with acromegaly is uniquely debilitating and may not be exclusively caused by pressure effects.
The systemic clinical features of acromegaly occur as a consequence of the deleterious impact of elevated serum concentrations of both GH and IGF-1 on peripheral tissues (Table 14-5). The somatic impact of elevated GH includes growth stimulation of a variety of tissues, such as skin, connective tissue, cartilage, bone, and many epithelial tissues, including mucosal surfaces. The metabolic effects of excess GH include nitrogen retention, insulin antagonism, and enhanced lipolysis.6
Table 14-5. Risk of Long-Term Exposure to Elevated Growth-Hormone Levels
GH, Growth hormone.
From Melmed S, Dowling RH, Frohman L, et al: Acromegaly: consensus for cure, Am J Med 97:468, 1994.
The onset of acromegaly is insidious, and disease progression is usually slow. At diagnosis, about 75% of patients are shown to harbor macroadenomas (tumor diameter of ≥10 mm), and some tumors extend to the parasellar or suprasellar regions.15 Headaches are the initial symptom in approximately 60% of patients, and 10% have visual symptoms.
Acral Overgrowth
Acral and soft-tissue overgrowth is invariably a feature of acromegaly. Characteristic findings include an enlarged, protruding jaw (macrognathia) with associated mandibular overbite and enlarged, swollen hands and feet, resulting in increasing shoe and glove size and the need to enlarge rings. Facial features are coarse, with enlargement of the nose and frontal bones, as well as the jaw; the upper incisors are consequently spread apart. Despite the prominence of these findings, the rate of change is so slow that few patients seek care because their appearance has changed (e.g., only 13% of 256 patients in one series5).
Rheumatologic Features
Musculoskeletal symptoms are leading causes of morbidity and serious functional disability in patients with acromegaly.96,97 In several studies encompassing large series, at least half of all patients exhibited minor arthralgias, and severe, debilitating arthritic features ultimately developed in more than one third of patients.97,98 The pathogenesis of joint disease in acromegaly generally begins with a noninflammatory osteoarthritic disorder and culminates in severe secondary joint and cartilage degeneration.97 Excess GH and IGF-1 exposure leads to uneven cartilage proliferation that results in a mechanically unstable joint surface. Joint spaces then narrow as weight-bearing surfaces erode cartilage and cause excess intraarticular new fibrocartilage deposits. Subchondral cysts and osteophytes then develop in an irreversible self-perpetuating process. Severe physical deformity and functional disability result from these inexorable pathologic and mechanical stresses. Although symptomatic and functional relief of arthritic disorders is observed in most patients after reducing GH levels, structural changes are unfortunately not reversible.98,99
Joint arthralgias are a common initial feature of the disease, and back pain and kyphosis are common.5 Synovial tissue and cartilage enlarge and cause hypertrophic arthropathy of the knees, ankles, hips, spine, and other joints.97 Back pain also may occur because of osteoporosis caused either by GH excess itself or concurrent gonadal insufficiency from the enlarging pituitary tumor. Spine and hip bone density may be increased in women with acromegaly, but not if estrogen deficiency is present.100 When excess GH secretion begins before epiphyseal fusion, linear growth increases and causes pituitary gigantism.
Skin and Soft Tissues
The skin thickens, and multiple recurrent skin tags may appear.3,101 Hyperhidrosis at rest is common (present in 50% of patients)2,3,5 and often malodorous. Hair growth increases, and some women have hirsutism.2,5,102 Other manifestations of soft-tissue overgrowth include macroglossia, deepening of the voice, and paresthesias of the hands (carpal tunnel syndrome) from nerve entrapment.2,5–7,15,98 Other patients have a symmetric sensorimotor peripheral (rarely hypertrophic) neuropathy unrelated to entrapment.96
Thyroid
Thyroid enlargement may be diffuse or multinodular. In a study of 37 patients with acromegaly, 92% had an enlarged thyroid gland when assessed with ultrasound; mean thyroid size was increased more than five times normal.103 Thyroid function is, however, usually normal.
Cardiovascular
Impaired cardiovascular function in acromegaly is an important determinant of morbidity and mortality,3,104 with exacerbated cardiovascular risk factors.198 The deleterious direct impact of excess GH and IGF-1 on the heart, as well as the effect of hypertension, which is present in 30% of patients, contributes to the disorder.5,105,106 Cardiac enlargement is disproportionate to the increased size of internal body organs,107–109 and the severity of cardiomyopathy correlates significantly with the duration of exposure to hypersomatotropism.104,108,110 Mean left ventricular mass may be significantly increased to more than 200 g, as opposed to a normal mean weight of 140 g, and end-systolic and diastolic volumes are attenuated. Concentric ventricular hypertrophy is associated with interstitial fibrosis, lymphocytic infiltration, and necrosis.108 Resting diastolic blood pressure and left and right ventricular peak filling rates are elevated. Post-exercise systolic and diastolic blood pressure may also be elevated, and the left ventricular ejection fraction is attenuated.111 Because physiologic doses of replacement GH also may actually improve cardiac function in patients with adult GH deficiency, a fine equilibrium may exist for the respective impacts of GH excess and GH deficiency on maintaining healthy myocardial function.108
Sleep Apnea
Peripheral airway obstruction caused by macroglossia, mandible deformation, mucosal hypertrophy, and inspirational laryngeal collapse has long been recognized as causing airway obstruction,5 snoring,3 and sleep apnea. Sleep apnea afflicts most patients with acromegaly and has been documented in up to 80% of cases.3 Macroglossia and enlargement of the soft tissues of the pharynx and larynx lead to obstructive sleep apnea in about 50% of patients; others have central sleep apnea, possibly resulting from altered central respiratory control.112 Sleep apnea may be an important cause of mortality in these patients. A central form of sleep apnea was recognized in acromegaly,112–114 which appears to correlate more closely with elevated GH and IGF-1 levels and may reflect central respiratory suppression caused by the dysregulated hypothalamic-GH axis. Clearly, the strong association of sleep apnea with hypertension, coronary artery disease, and cardiac arrest also reflects the clinical phenotype of patients with acromegaly. Attenuation of GH levels, especially with octreotide, improves or abrogates sleep apnea.112,115 Octreotide treatment is associated with improved indices for apnea, hypopnea, and oximetry.195 After 6 months of treatment of 14 apneic acromegalic patients with octreotide, a 40% decrease in the number of apneic events per hour was seen, as well as a decrease in total apneic time from 28% to 15%. Maximum O2 saturation rose from 76% to 84%, accompanied by a decline in daytime sleepiness, as well as improvement in central and obstructive apneic parameters.112
Diabetes
GH is a potent antagonist of insulin action, and glucose intolerance is encountered in up to 60% of patients. About 25% of patients may require insulin, and thus diabetes is an important systemic complication of hypersomatotropism. Diabetes is a major determinant of mortality, and only 30% of patients with diabetes at the time acromegaly is diagnosed appear to survive 20 years.83,116
Gonadal Function
Women with acromegaly may have amenorrhea, with or without galactorrhea,3,15,117 and some have hot flashes and vaginal atrophy. Men may have impotence, loss of libido, decreased facial hair growth, and testicular atrophy.3,7,117 Hypogonadism is caused either by hyperprolactinemia (present in about 30% of patients)102,117 or by impairment of gonadotropin secretion as the expanding pituitary tumor compresses normal pituitary gonadotroph cells. Asymptomatic, reversible prostatic enlargement also is common, even in men with hypogonadism.118,119
Neoplasms
Acromegaly is associated with an enhanced risk for development of colonic polyps,120–122 and prospective studies have reported premalignant adenomatous colonic polyps in up to 30% of patients, a prevalence not different from that in the general U.S. population.122–124 In patients with acromegaly, colon length is increased, and apoptotic activity is decreased significantly.121,209 Patients with acromegaly are more likely to have multiple adenomatous polyps, as well as polyps proximal to the splenic flexure, underscoring the need for full-length colonoscopy.121,125 No difference in the duration or degree of acromegaly is evident in patients with or without adenomatous polyps. A multicenter retrospective study of 1362 patients with acromegaly found a lower cancer rate than in the general population (standardized incidence ratio of 0.76) but an increased colon cancer mortality rate.126,127 The enhanced mortality correlated with persistently elevated serum GH concentrations but was not observed in patients with posttreatment serum GH levels less than 2.5 ng/mL.128,129 Overall, a recent meta-analysis suggests a moderate doubling of colon cancer risk in acromegaly.210 Colonoscopy is therefore recommended at diagnosis for all patients and periodically thereafter. Uncontrolled GH levels may act permissively to enhance morbidity and mortality from colon cancer. These findings underscore the requirement for tight GH control in these patients.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


