FIGURE 41-1. Summary of the metabolic abnormalities in type 2 diabetes mellitus that contribute to hyperglycemia. Increased hepatic glucose production, impaired insulin secretion, and insulin resistance caused by receptor and postreceptor defects all combine to generate the hyperglycemic state.
Although the causal mechanisms may be heterogeneous in different type 2 diabetic patient groups, ultimate expression of the hyperglycemic state involves some combination of impaired insulin secretion, insulin resistance, and increased hepatic glucose production, and the relative magnitude and importance of these three common metabolic abnormalities depend on the specific genetic, pathologic, or environmental factors involved in a particular patient.
Genetic versus Acquired Factors
GENETIC FACTORS
Abundant evidence supports the view that a strong genetic component contributes to type 2 diabetes. Although many patients have a positive family history for this disease, perhaps the strongest evidence comes from twin studies. In one study, 53 twin pairs were examined, in whom one twin was ascertained to have type 2 diabetes. On assessment of the other twin, type 2 diabetes had developed in 91% (48/53) of the co-twins.15 Although the five discordant twins were not overtly diabetic, they had mild glucose intolerance and abnormal insulin responses during oral glucose tolerance tests, suggesting that they too might ultimately progress to overt disease.
Further evidence for a genetic basis comes from striking differences in the prevalence of type 2 diabetes in various ethnic groups that are not explained by environmental factors. The prevalence of type 2 diabetes in the United States is 2% to 4% for Caucasians, but it is 4% to 6% for African Americans16 and 10% to 15% for Mexican Americans,17 and these numbers are increasing as the prevalence of obesity is rising at epidemic rates. More than 40% of the Pima Indians in Arizona have type 2 diabetes mellitus; this is the group with the highest incidence of type 2 diabetes in the world.18 Among the Pima Indians, 80% of 35- to 44-year-old offspring of two parents with type 2 diabetes mellitus before age 45 years have diabetes, and a positive family history of type 2 diabetes is a substantial risk factor for disease development. Clearly, this genetic predisposition interacts with adverse environmental influences, such as obesity and sedentary lifestyle, which are largely responsible for the sharp uptick in type 2 diabetes mellitus incidence in recent years.
Despite the high concordance rate in twins, “garden variety” type 2 diabetes is obviously not simply the result of a single-gene defect; the inheritance pattern of type 2 diabetes does not conform to any recognizable mendelian pattern, and the incidence of type 2 diabetes in first-degree relatives is below what one would expect. In most patients with type 2 diabetes, the disease appears to be a polygenic disorder, in that disease expression depends on multiple gene loci, all of which may have small to moderate effects. It also is likely that type 2 diabetes is multigenic, meaning that different combinations of gene polymorphisms may exist among patients. A more detailed discussion of possible susceptibility genes will follow later in this chapter. Type 2 diabetes is referred to as a multifactorial disease, in that the genes interact not only with each other, but also with environmental influences. Individuals therefore may be predisposed to develop type 2 diabetes through their inheritance of a particular combination of genes, but acquired environmental factors are necessary to bring out the phenotypic manifestation of hyperglycemia.
Although multiple genes are involved in the pathogenesis of diabetes in most patients, a small number of patients have a monogenic form of this disease. In these forms of diabetes, environmental factors play little or no role in determining the phenotypic expression. These single-gene mutations may confer a defect in insulin secretion or insulin action, and each will be discussed specifically later in the chapter.
ACQUIRED FACTORS
Lifestyle: Diet, Exercise, and Obesity
Acquired factors play a major role in the development of type 2 diabetes in genetically predisposed individuals; this is clearly demonstrated by assessment of the impact of lifestyle changes on prevalence of diabetes in various ethnic populations. The prevalence of diabetes increases as ethnic groups migrate from lesser developed to more urbanized areas or simply change from an agrarian to a more sedentary, urban lifestyle. The former has been illustrated by surveys in Japanese subjects. In rural Japan, the prevalence of type 2 diabetes was approximately 4%,19 whereas among Japanese who have immigrated to the United States, the prevalence rises to more than 21%.20 This is also the case in the Pima Indians, who in Arizona have adopted a largely “Westernized” lifestyle, although those living in northwestern Mexico have remained agrarian. The Indians in Arizona have a prevalence of diabetes of 54% and 37% for men and women, respectively, whereas the Mexican Indians have a prevalence of 6% and 11%, respectively.21
It is likely that nutrition and lifestyle are the environmental factors that explain the difference in diabetes prevalence in genetically similar populations. Adoption of an urbanized, Westernized lifestyle is associated with change to a diet that has a higher content of total calories, fats, and refined carbohydrates. For example, the mean daily intake of fat among Japanese men living in Japan was reported to be 16.7 g; by contrast, in Japanese American men, the mean intake was 32.4 g.22 In addition, the level of physical activity is lower among ethnic groups living in the United States compared with the same ethnic groups living in their country of origin.23 These lifestyle changes obviously predispose to the development of obesity, and overwhelming evidence suggests that obesity is a major factor in the development of diabetes. The role of obesity in the pathogenesis of diabetes will be discussed in detail later in this chapter. Recent evidence supporting the role of lifestyle factors in the development of diabetes has come from the Diabetes Prevention Program.24 In this study, intensive lifestyle modification, consisting of dietary change and increased exercise, led to a 58% reduction in the progression of impaired glucose tolerance to diabetes over a 2.8 year period.
The extent of the contribution of these lifestyle factors, independent of their association with obesity, to the development of diabetes remains unclear. For example, it is not known whether specific dietary components, such as a diet that is rich in saturated fat or highly refined carbohydrates, play an independent role in the pathogenesis of type 2 diabetes. With regard to exercise, however, evidence from experimental and epidemiologic studies indicates that subjects with lower physical activity levels are more prone to develop diabetes, independent of obesity.25
Low Birth Weight
Low birth weight as a risk factor for the development of insulin resistance and diabetes mellitus later in life has been described in many populations over the past 2 decades.26–28 The mechanisms responsible for this association are unknown but may be related to epigenetic fetal adaptation to nutritional stimuli or excess fetal glucocorticoid exposure.29
Aging
Aging is associated with a decrease in glucose tolerance, which appears to be due to a decline in both insulin sensitivity and insulin secretion.30 However, age-related factors such as reduced physical activity and increased fat accumulation are at least in part responsible for this phenomenon. Obviously, type 2 diabetes incidence increases with age, but whether the aging process per se is contributory remains unclear.
In summary, in most subjects, type 2 diabetes is a heterogeneous disorder that results from a complex interaction between genetics and environmental influences such as obesity and other acquired factors.
Natural History of Type 2 Diabetes
The pathophysiologic findings depicted in Fig. 41-1 represent a single point in time after overt type 2 diabetes has developed. However, such an analysis does not reveal the progressive evolution of this disease. Fig. 41-2 presents a schematic description of the natural history or progression to type 2 diabetes. Evidence indicates that in most populations, those who evolve to type 2 diabetes begin with insulin resistance. Insulin resistance can be a primary inherited feature, but acquired factors such as obesity, sedentary lifestyle, and aging (particularly obesity) also can be causal or can exacerbate underlying genetic mechanisms of decreased insulin sensitivity. In an attempt to overcome insulin resistance, the β cell increases insulin secretion, resulting in hyperinsulinemia, which is able to maintain relatively normal glucose tolerance. In a subpopulation of subjects, however, this hyperinsulinemic response is insufficient to fully compensate for the prevailing insulin resistance, and impaired glucose tolerance (IGT) develops. Although a percentage of subjects with IGT may revert to normal glucose tolerance, IGT should be considered an intermediate stage in the development of type 2 diabetes, with many subjects eventually progressing to frank expression of the disease.
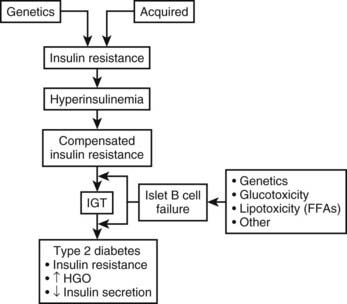
The proportion of IGT subjects who progress to type 2 diabetes depends on the particular ethnic groups studied and the methods of assessment used. For example, in the National Institutes of Health (NIH)-sponsored multicenter Diabetes Prevention Program, ≈10% of patients with IGT developed type 2 diabetes mellitus per year.31 During the transition from IGT to frank type 2 diabetes, at least three pathophysiologic changes can be observed. First is a marked fall in β cell function and insulin secretion. Whether this decrease is due to preprogrammed genetic abnormalities in β cell function, to acquired defects (such as glucotoxicity or lipotoxicity), or to both remains to be elucidated. Nevertheless, a marked decrease in β cell function accompanies this transition, and most believe that this decreased β cell function is the major contributor to the progression to type 2 diabetes mellitus. A second metabolic change is seen at the level of the liver. Subjects with IGT have normal basal rates of hepatic glucose output (HGO), whereas patients with fasting hyperglycemia have increased HGO. Thus, the capacity of the liver to overproduce glucose is an important contributory factor (albeit secondary) to the pathogenesis of type 2 diabetes. Finally, many but not all studies have indicated that patients with type 2 diabetes are more insulin resistant than are those with IGT. Most likely, this increase in insulin resistance is secondary to glucotoxicity or to other acquired factors.
Evidence implicating insulin resistance as a primary defect comes principally from studies examining subjects who are at increased risk for developing diabetes. One such group consists of individuals whose parents have type 2 diabetes. Using intravenous glucose tolerance tests (GTTs), Warram and coworkers evaluated 155 nondiabetic offspring whose parents both had type 2 diabetes.32 During a follow-up period averaging 13 years, type 2 diabetes developed in 16% of the total group. However, when offspring were categorized on the basis of insulin sensitivity at initial testing, the cumulative incidence of diabetes was 60% among subjects with preexisting insulin resistance and less than 5% in insulin-sensitive offspring. Thus, insulin resistance and hyperinsulinemia (rather than hypoinsulinemia) characterized the prediabetic state, and this occurred irrespective of obesity and antedated by many years the subsequent development of impaired insulin secretion and overt type 2 diabetes.32
Studies in ethnic populations with a high prevalence of type 2 diabetes also have provided data supporting a primary role for insulin resistance in diabetes development. Thus, among Pima Indians, hyperinsulinemia and an associated decrease in insulin-mediated glucose disposal are early abnormalities that predict the subsequent development of both IGT and type 2 diabetes.33,34 Pima Indians with IGT who progress to type 2 diabetes have lower insulin levels 2 hours after a glucose load than do those who continue to have IGT or who return to normal glucose tolerance. Similar results come from studies of Micronesians in Nauru, a population with a prevalence of type 2 diabetes of approximately 30%.35 Again, in this population, IGT and type 2 diabetes were most likely to develop in those with hyperinsulinemia at baseline, but progression from IGT to type 2 diabetes could be predicted by lower baseline insulin responsiveness to a glucose challenge.
Further evidence comes from studies in first-degree relatives of type 2 diabetic patients.36–38 In these studies, peripheral insulin resistance and hyperinsulinemia were found in normoglycemic relatives of diabetic patients. First-degree relatives with IGT displayed insulin resistance but also exhibited defects in insulin secretion that were not apparent in those with normal glucose tolerance.
In summary, the phenotypic manifestation of type 2 diabetes mellitus involves elevated HGO, impaired insulin secretion, and peripheral insulin resistance. Type 2 diabetes mellitus has a strong genetic component, and studies of prediabetic subjects indicate that in most populations, insulin resistance, accompanied by hyperinsulinemia, exists before any deterioration in glucose homeostasis occurs. After a period of compensatory hyperinsulinemia with normal glucose tolerance, β cell insulin secretion declines, and IGT and eventually overt type 2 diabetes mellitus result.
In the remainder of this chapter, abnormalities of insulin secretion, insulin action, and hepatic glucose metabolism in type 2 diabetes are reviewed, with particular attention to basic mechanisms (where known) and their applicability to etiology.
Pathophysiology of Type 2 Diabetes Mellitus
ABNORMAL PANCREATIC β CELL FUNCTION
Obesity is a major cause of insulin resistance, and as was described earlier, the β cell compensates for decreased insulin sensitivity by increasing insulin secretion. In normal glucose-tolerant subjects, the increase in insulin secretion that occurs with insulin resistance is described by a hyperbolic relationship39 (Fig. 41-3). Although this quantitative increase in insulin secretion in response to decreased insulin sensitivity should be viewed as an appropriate response, subtle qualitative changes in insulin secretion also are noted in the insulin-resistant state. Insulin normally is secreted in rapid regular pulses with a 5 to 15 minute frequency superimposed on slower ultradian oscillations every 80 to 150 minutes.40 These normal rapid, regular pulses are replaced by disordered pulses in obese subjects with insulin resistance and also in the insulin-resistant but glucose-tolerant offspring of subjects with type 2 diabetes.41 Indeed, insulin secretory pulse frequency has been shown to correlate inversely with peripheral insulin sensitivity.42,43
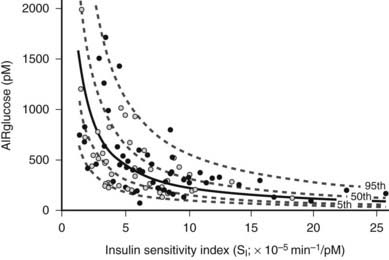
FIGURE 41-3. Relationship between insulin sensitivity and β cell function quantified as the first-phase insulin response (AIRglucose) in 93 (55 males and 38 females) apparently healthy, nondiabetic subjects younger than 45 years. The cohort demonstrates a broad range of insulin sensitivity and β cell function. The solid curve depicts the best fit relationship (50th percentile); the dashed curves represent the 5th, 25th, 75th, and 95th percentiles. The relationship is best described by a hyperbolic function, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in β cell function.
(Data from Kahn SE, Prigeon RL, McCulloch DK, et al: Quantification of the relationship between insulin sensitivity and β cell function in human subjects: evidence for a hyperbolic function, Diabetes 42:1663–1672, 1993.)
Insulin Secretion in Subjects With Impaired Glucose Tolerance
In subjects with IGT, both quantitative and qualitative defects in insulin secretion are usually present, although this can be variable.43,44 Part of this variability may be explained by the heterogeneity of this condition, as some subjects with IGT will revert to normal glucose tolerance, a proportion will progress to frank type 2 diabetes, and others will continue to have IGT for many years.45
With regard to the hyperbolic relationship between β cell function and insulin sensitivity, subjects with IGT secrete less insulin than is appropriate for their degree of insulin resistance.46 Furthermore, using the graded glucose infusion method, Polonsky has shown that subjects with IGT secrete less insulin at any given glucose level than do normoglycemic subjects matched for a similar degree of insulin resistance and obesity (Fig. 41-4).47 Insulin-resistant subjects with IGT who have progressive impairment in insulin secretion are most likely to develop full-blown type 2 diabetes.48
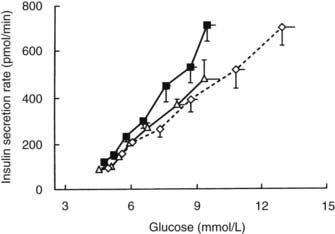
FIGURE 41-4. Relationship between average plasma glucose concentrations and insulin secretion rates during graded glucose infusion studies in a group of lean nondiabetic control subjects (open triangles), nondiabetic obese subjects (closed squares), and matched obese subjects with impaired glucose tolerance (open diamonds). The lowest glucose levels and insulin secretion rates were measured under basal conditions, and subsequent levels were obtained during glucose infusion rates of 1, 2, 3, 4, 6, and 8 mg/kg/min. Values are means ± standard error of the mean (SEM).
(Data from Polonsky KS: The β-cell in diabetes: from molecular genetics to clinical research, Diabetes 44:705–717, 1995.)
Insulin secretion in response to a sustained intravenous glucose stimulus is normally biphasic: A rapid rise in insulin levels within 1 to 3 minutes (first phase) is followed by a return to baseline within 6 to 10 minutes with a subsequent gradual increase (second phase). Subjects with IGT have a reduction in both first- and second-phase responses49 to glucose, and a further qualitative defect in insulin secretion in IGT is the replacement of rapid regular secretory oscillations with disorganized pulses.41
Insulin Secretion in Subjects With Type 2 Diabetes
The abnormalities of insulin secretion described in subjects with IGT are also present in type 2 diabetes, although to a more marked degree. β cell function progressively deteriorates during the natural history of type 2 diabetes. This decline in β cell function is evident not only during the conversion from compensated insulin resistance to IGT and subsequently to overt type 2 diabetes, but also during the progressive course of established type 2 diabetes after its initial onset.50
Basal insulin levels usually are normal or increased in type 2 diabetes. Indeed, obese subjects with type 2 diabetes can have basal insulin levels severalfold higher than normal, but this does not mean that basal β cell secretory function is normal, because the prevailing plasma glucose level also must be taken into account.51–53 Hyperglycemia is the major stimulus for insulin secretion, and when normal individuals are made hyperglycemic by infusion of glucose, circulating insulin levels are much higher than those found in type 2 diabetes.51,52 Thus, patients with type 2 diabetes maintain normal or increased basal insulin levels only in the face of the enhanced stimulus of fasting hyperglycemia, which indicates an underlying impairment in the sensitivity of the β cell to glucose. Stimulated insulin levels in type 2 diabetes can be low, normal, or high depending on factors such as the severity of diabetes, the degree of obesity, and the preceding level of diabetic control.13,14,44
In Response to Intravenous Glucose
In type 2 diabetes, defects in the insulin secretory response to intravenous glucose are observed consistently. Once fasting plasma glucose levels exceed 126 mg/dL, the first-phase insulin response to intravenous glucose characteristically is completely absent. This relationship is shown in Fig. 41-5.1,54,55
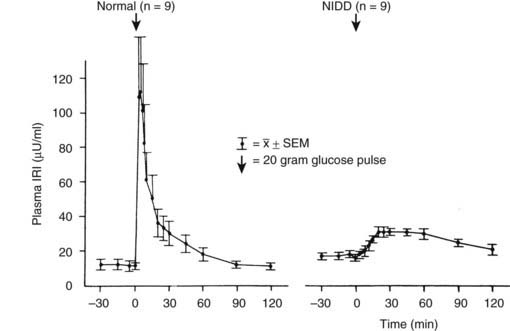
FIGURE 41-5. First-phase insulin release in response to the intravenous administration of glucose in normal and type 2 diabetic (non–insulin-dependent diabetes [NIDD]) subjects. Mean fasting plasma glucose concentrations were 83 ± 3 mg/dL in normal subjects and 160 ± 10 mg/dL in type 2 diabetic subjects.
(Data from the American Diabetes Association, Inc., from Ward WWK, Beard JC, Halter JB, et al: Pathophysiology of insulin secretion in non-insulin-dependent diabetes mellitus, Diabetes Care 7:491–502, 1984.)
It is interesting to note that acute or first-phase insulin secretion in response to nonglucose stimuli such as arginine1 or isoproterenol56 is relatively preserved; this finding indicates a functionally selective β cell defect in response to glucose stimuli in type 2 diabetes. Because the acute insulin response to intravenously administered arginine or isoproterenol increases as the glucose concentration is raised, Porte and colleagues have attempted to quantitate this effect of glucose by plotting the increase in acute insulin response to arginine or isoproterenol pulses as a function of increasing plasma glucose level.1,14,51 The slope of this relationship is termed the glucose potentiation slope, and by this analysis, glucose potentiation of β cell function is also reduced in type 2 diabetes.1,14
Although absent first-phase insulin secretion may be a marker for β cell dysfunction, it is unlikely to be an important cause of glucose intolerance or hyperglycemia. Thus, mildly hyperglycemic and severely hyperglycemic type 2 diabetic patients are equally deficient in first-phase insulin secretion, implying that this deficiency does not play a role in further deterioration in glucose tolerance from mild to severe fasting hyperglycemia. Furthermore, in selected patients with normal glucose tolerance in whom type 1 diabetes eventually develops, the acute insulin response to intravenous glucose is absent during the normal stage and therefore does not cause hyperglycemia.57 Finally, α-adrenergic blockers can substantially restore the acute insulin response to intravenous glucose58 without major improvement in fasting glycemia or glucose tolerance.
Second-phase insulin secretion, which is assessed with the hyperglycemic clamp or the graded intravenous glucose infusion technique (see Fig. 41-4 for details), is markedly reduced in individuals with type 2 diabetes compared with normal subjects and those with IGT. In general, the more severe the diabetes, the lower is the second-phase insulin response.1,54
In Response to Oral Glucose and Mixed Meals
The insulin response in type 2 diabetes to oral ingestion of glucose or mixed meals is far more variable than the response to intravenous glucose. After oral glucose, insulin levels are usually subnormal in type 2 diabetes,14,36 although this might not always be the case in patients with mild hyperglycemia.44 This heterogeneity is depicted in Fig. 41-6, which summarizes the results of oral GTTs in a wide spectrum of normal and type 2 diabetic subjects.59 As can be seen, hyperinsulinemia frequently exists in mild states of glucose intolerance. In individuals with mild diabetes, insulin levels generally are in the “normal range,” although inappropriately or relatively low for the degree of hyperglycemia and insulin sensitivity. With more severe diabetes, absolute stimulated insulin levels are uniformly low.
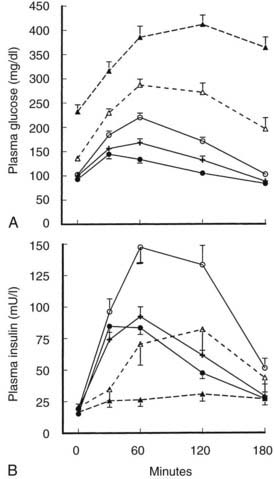
FIGURE 41-6. A, Mean (±standard error of the mean [SEM) plasma glucose response to oral glucose in the five subject groups. Closed circle, normal; cross, borderline tolerance; open circle, impaired glucose tolerance; open triangle and broken line, fasting hyperglycemia (110 to 150 mg/dL); closed triangle and broken line, fasting hyperglycemia (>150 mg/dL). B, Mean (±SEM) plasma insulin response to oral glucose in the five subject groups. Symbols are the same as in A.
(Data from Reaven GM, Olefsky JM: Relationship between heterogeneity of insulin responses and insulin resistance in normal subjects, Diabetologia 13:201–206, 1977.)
In type 2 diabetes, the defect in β cell function is relatively (but not completely) specific for glucose stimuli. The relative preservation of insulin responses to nonglucose stimuli, such as certain amino acids and the incretin gut peptides, means that type 2 diabetic patients have a better insulin response to mixed meals than to oral glucose. Thus, in mild type 2 diabetes, insulin responses to mixed meals may be delayed, but postprandial hyperglycemia, coupled with other insulinogenic factors, often leads to exaggerated insulin responses 2 to 4 hours after meal ingestion. However, with more severe hyperglycemia, decreased insulin levels are more common. Because oral GTTs represent a rather nonphysiologic stress by which to assess β cell function, it is important to keep in mind that in the free-living state, when patients are consuming mixed meals, absolute insulin levels can be relatively preserved in type 2 diabetes.
Proinsulin Secretion
Another factor related to hyperinsulinemia in type 2 diabetes is circulating proinsulin levels. Proinsulin is secreted by β cells concomitantly with insulin and cross-reacts with insulin in most insulin immunoassays, thus contributing to the total measured immunoreactive insulin level. In normal subjects, proinsulin represents only a small portion (3% to 7%) of the insulin-like material secreted by β cells. However, using proinsulin-specific immunoassays, several groups have shown that in hyperinsulinemic states and in many cases of type 2 diabetes, an increased proportion of proinsulin is released and contributes to the measured insulin in standard immunoassays, so true insulin levels are overestimated.14,53,60 When corrected for this factor, basal insulin levels may be normal or moderately elevated in type 2 diabetes.
Mechanisms of β Cell Dysfunction
The precise cellular mechanisms underlying β cell dysfunction in type 2 diabetes are unclear, but they most likely represent a combination of genetic and acquired factors.
The overall mass of β cells changes in obesity and type 2 diabetes.61,62 β cell mass reflects the balance between new islet formation (neogenesis) and β cell loss due to apoptosis. Longitudinal studies in animal models suggest that β cell mass increases appropriately in response to a decrease in insulin sensitivity,62 and cross-sectional data from humans have long revealed an expanded β cell mass in obesity.63 In contrast, β cell mass is reduced in type 2 diabetes.61 On the basis of murine models and cross-sectional autopsy data from humans, diminished β cell mass is thought to be due to accelerated β cell apoptosis and the failure of islet neogenesis and β cell replication to compensate for this loss.62
It has been theorized that lipid accumulation in the β cell is implicated in the apoptotic process and the development of impaired insulin secretion in type 2 diabetes. This is referred to as lipotoxicity and may involve excess fatty acids entering β cells, thus triggering the apoptotic cellular response.64
A body of evidence also suggests a role for islet amyloid polypeptide (IAPP) in the loss of β cells. IAPP is synthesized in the β cell and is co-secreted with insulin.65 IAPP aggregates to form fibrils of amyloid, and islet amyloid is found at autopsy in up to 90% of subjects with type 2 diabetes.66 In vitro, IAPP is toxic, causing β cell apoptosis,67 and it may contribute to the reduced β cell mass that is associated with type 2 diabetes.
Insulin secretory abnormalities found in type 2 diabetes are often improved after a period of good blood glucose control, irrespective of the treatment used (diet, insulin, or oral hypoglycemic agents).68–70 This partial reversibility is consistent with the idea that, to some extent, the abnormalities may be secondary to hyperglycemia or some other factor associated with uncontrolled diabetes. Support for the “glucotoxicity” theory comes from a variety of in vivo and in vitro studies showing that chronic exposure of islets to hyperglycemia can result in a number of different defects in glucose-induced insulin secretion.71 It is important to note that when isolated human islets are incubated under euglycemic and hyperglycemic conditions, islets that are exposed to hyperglycemia demonstrate a marked defect in their ability to secrete insulin in response to subsequent glucose stimuli.72 Although the precise mechanism is unknown, it seems likely that glucotoxicity coupled with lipotoxicity plays some role in the impaired β cell function of type 2 diabetes.
An interesting finding from studies of mouse genetics is that insulin receptor signaling in β cells is important for normal function. Thus, mice in which the insulin receptor gene has been specifically deleted from β cells show a complete loss of first-phase insulin secretion in response to glucose but not arginine, reminiscent of the β cell defect in type 2 diabetes.73 Second-phase glucose-induced insulin secretion is also blunted in these mice, and they show an age-dependent progressive impairment in glucose tolerance. Glucose-stimulated insulin secretion involves transport of glucose into cells by a specific glucose transporter, termed GLUT2. Following uptake, glucose is phosphorylated by glucokinase, and subsequent intracellular metabolism of glucose-6-phosphate leads to stimulation of insulin secretion. In mouse studies, genetic deletion of GLUT2 leads to loss of glucose but not arginine-stimulated insulin secretion. It is interesting to note that feeding mice high-fat diets to induce obesity also leads to a decrease in β cell GLUT2 expression, suggesting another mechanism of interaction between acquired environmental factors and β cell dysfunction.
The decrease in β cell mass in type 2 diabetes is in the range of 30% to 50%. However, because sufficient insulin secretory reserve normally exists to sustain an 80% to 90% loss of β cells without the development of hyperglycemia, it follows that decreased functional capacity of the remaining β cells must exist in type 2 diabetes. Indeed, it has been shown that the maximal insulin secretory capacity may be reduced by as much as 80% in type 2 diabetic subjects.52 It is possible that the decrease in β cell mass in type 2 diabetes is somehow causally related to the decreased function of the remaining β cells. Thus, partially pancreatectomized rats and streptozotocin-treated rats display similar insulin secretory defects,71 which suggests that decreased glucose-stimulated insulin secretion with relative preservation of responsiveness to nonglucose stimuli may be a general type of abnormality that may occur in response to a variety of β cell insults.
PERIPHERAL INSULIN RESISTANCE
In addition to impaired β cell function, another primary pathophysiologic disorder underlying type 2 diabetes mellitus is insulin resistance. Insulin resistance is a metabolic state in which a normal concentration of insulin produces a less than normal biological response. This decreased response to insulin can involve any of the multiple metabolic effects of insulin, but from the standpoint of relevance to type 2 diabetes, resistance to the effects of insulin on glucose metabolism has been the most extensively studied. Because insulin travels from the β cell through the circulation to the target tissue, events at any of these loci can influence the ultimate action of the hormone.
Causes of Peripheral Insulin Resistance
Circulating Factors That Influence Insulin Action
Hormonal antagonists include all known counterregulatory hormones such as cortisol, growth hormone, glucagon, and catecholamines. In well-known syndromes (e.g., Cushing’s disease, acromegaly), elevated levels of these hormones can induce an insulin-resistant diabetic state. However, in the usual case of obesity or type 2 diabetes, excessive levels of these counterregulatory hormones are not an important contributory factor to insulin resistance.
As the body’s largest endocrine organ, adipocytes secrete a number of polypeptides, called adipokines, into the circulation. In turn, these adipokines (i.e., adiponectin, leptin, resistin, retinal binding protein 4, and others) exert distal effects on glucose homeostasis in an endocrine fashion. The effects of adipokines will be discussed later in the chapter.
Several years ago, Randle and coworkers hypothesized that the elevated circulating levels of free fatty acids (FFAs) found in obesity and type 2 diabetes impair peripheral glucose utilization.74 Substantial evidence indicates that FFAs do indeed contribute to insulin resistance, although the mechanisms differ from those originally proposed by Randle. FFAs also play an important role in the regulation of HGO and contribute to hepatic insulin insensitivity in obesity and type 2 diabetes. These mechanisms will be discussed in greater detail later in this chapter.
Other circulating antagonists, such as antibodies against the insulin molecule or against the insulin receptor, are rare causes of insulin resistance and are discussed later in the chapter under “Syndromes of Severe Insulin Resistance.”
Impaired Access of Insulin to Target Cells
Because insulin must travel from the circulation to target tissues to elicit biological effects, any defect in this transfer could lead to functional insulin resistance. Compared with secretion into the circulation, the passage of insulin from the plasma compartment to tissue sites of action is markedly delayed, and in vivo effects of insulin in stimulating glucose disposal are well correlated with the appearance of insulin in the interstitial fluid.75 Lymph and interstitial insulin levels are ≈40% lower than those in plasma,75,76 which indicates that peripheral tissues are more sensitive to insulin than was previously recognized. Furthermore, the possibility arises that either the rate or the amount of insulin being transferred from the plasma to the interstitial compartment could be abnormal in type 2 diabetes or obesity, thereby contributing to the insulin-resistant state.77,78 Recent studies indicate that transport of insulin across the capillary in vivo occurs by diffusion76 and is not receptor mediated, as was previously suggested.79 Transport by diffusion fits with the finding that transcapillary passage is comparable in normal subjects, insulin-resistant nondiabetic subjects,80 and those with type 2 diabetes.81 Further evidence that the delayed activation of muscle glucose uptake in obesity and type 2 diabetes is not due to impaired transcapillary transport of insulin comes from a study by Nolan and coworkers.78 This study showed that the kinetic defect in the ability of insulin to stimulate leg glucose uptake was not accompanied by any delay in the activation of leg muscle insulin receptors by insulin, thus implying that the kinetic defect is distal to the insulin receptor.
Another physical factor that may relate to insulin resistance is muscle capillary density, which correlates with in vivo insulin sensitivity.82 Laakso and coworkers have shown that insulin, at least at pharmacologic levels, increases leg blood flow.83 Because tissue glucose uptake is a product of blood flow and the arteriovenous glucose difference, increased leg blood flow could contribute to overall glucose disposal. Similar studies performed in obese subjects and in subjects with type 2 diabetes revealed a decrease in the insulin-induced increase in leg blood flow, which may explain part of the decrease in total leg glucose uptake.83 However, others found no effect of insulin on blood flow,84 and Utriainen and colleagues used 15O [H2O] and positron emission tomography to confirm enhancement of leg muscle blood flow by pharmacologic insulin levels, but no difference in the response between type 2 diabetic and normal control subjects.85
Although some of these factors might be contributory, they cannot explain the major component of insulin resistance in type 2 diabetes and obesity, because numerous studies have demonstrated profound in vitro insulin resistance in tissues and cells from these patients.
Cellular Defects in Insulin Action
Available evidence points to a target tissue defect as the major cause of insulin resistance in type 2 diabetes. Before potential causes are considered, it is useful to review some general concepts concerning normal insulin action (Fig. 41-7). Insulin first binds to its cell surface receptor, a heterotetrameric glycoprotein composed of two α subunits (135 kDa) and two β subunits (95 kDa) linked by disulfide bonds.86–89 The α subunits are entirely extracellular and are responsible for insulin binding. The β subunits are transmembrane proteins containing a small extracellular domain and a larger cytoplasmic domain that includes insulin-regulated tyrosine kinase activity. Binding of insulin to the receptor rapidly induces tyrosine autophosphorylation of the β subunit involving three tyrosine residues in the kinase domain, in addition to tyrosine residues adjacent to the transmembrane domain and in the C terminus of the β subunit. Once the receptor has been autophosphorylated, its intrinsic tyrosine kinase catalytic activity is markedly enhanced, and it now can phosphorylate tyrosine residues on endogenous protein substrates. Activation of the insulin receptor tyrosine kinase is essential for transduction of the insulin signal and for internalization of the receptor. Patients with naturally occurring mutations in the tyrosine kinase domain of the insulin receptor have syndromes of severe insulin resistance.
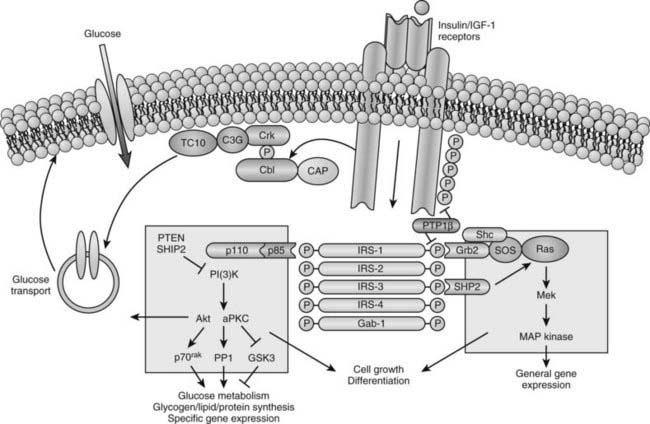
FIGURE 41-7. Model of cellular insulin action.
(From Saltiel AR, Kahn CR: Insulin signalling and the regulation of glucose and lipid metabolism, Nature 414:799–806, 2001.)
In recent years, major advances have been made in our understanding of how the insulin signal is propagated downstream from the activated insulin receptor to various insulin-regulated enzymes, transporters, and insulin-responsive genes to mediate its metabolic and growth effects (see Fig. 41-7). This field is rapidly evolving and complex and is discussed only briefly here because it is covered comprehensively in Chapter 34. A large number of intermediate signaling molecules have been identified, and after activation of the insulin receptor kinase, more than one signaling pathway may be used (see Fig. 41-7). For example, some of the components in the pathways leading to mitogenic effects of insulin are distinct from those leading to activation of glucose transport. Even a single action of insulin such as stimulation of glucose transport can involve more than one signaling pathway. Several cytosolic protein substrates of the insulin receptors are phosphorylated on tyrosine residues within seconds of insulin binding to its receptor. The first of these substrates to be identified was insulin receptor substrate 1 (IRS-1).89,90 IRS-1 belongs to a growing family of proteins that includes IRS-2, IRS-3, IRS-4, and a protein termed shc, which are immediate substrates of the insulin receptor kinase involved in insulin signaling. These proteins have no enzymatic activity but act as docking proteins. Tyrosine phosphorylation of these substrates enhances their association with proteins that contain src homology-2 (SH2) domains. These SH2 domains contain ≈100 amino acids and can bind to specific short motifs that encompass a phosphotyrosine. The binding of specific SH2 domain–containing proteins to tyrosine-phosphorylated IRS proteins or shc generates multicomponent signaling complexes, which, in turn, modulate the activities of phosphoinositide-3-kinase (PI3K), several serine kinases, and phosphatases that act on key insulin-regulated enzymes and transcription factors.
One of the most important effects of insulin with respect to type 2 diabetes is stimulation of glucose uptake into skeletal muscle, adipocytes, and heart muscle. Under most physiologic circumstances, glucose transport in these tissues is rate limiting for overall glucose disposal.91–93 Tissue glucose uptake is mediated by a family of at least five facilitative glucose transporters, each derived from a separate gene. These transporters show a high degree of homology, but each has tissue-specific distribution.94,95 One of them, GLUT4, or the insulin-sensitive glucose transporter, is uniquely expressed in skeletal muscle, adipose tissue, and cardiac muscle. In the unstimulated state, most of the GLUT4 proteins are located in an intracellular vesicular pool. Upon insulin stimulation, recruitment or translocation of these glucose transporter-rich vesicles to the cell surface causes insertion of GLUT4 proteins into the plasma membrane, where they begin to transport glucose into the cell.96–100
Clearly, insulin action involves a cascade of events, and abnormalities anywhere along this sequence can lead to insulin resistance.
Characteristics of Insulin Resistance in Subjects With Impaired Glucose Tolerance or Type 2 Diabetes
The frequency of insulin resistance increases as the degree of carbohydrate intolerance worsens.101 Thus, many, but not all, subjects with IGT are insulin resistant, whereas essentially every type 2 diabetic patient with fasting hyperglycemia displays this abnormality. Numerous studies indicate that insulin resistance is more marked in type 2 diabetes than in the prediabetic IGT state.3,5,36 However, other reports show only a modest increase in the degree of insulin resistance going from IGT to type 2 diabetes. Because most type 2 diabetic patients are overweight, obesity-induced insulin resistance is clearly a major contributing factor in these patients. However, obesity is not the only cause, in that the insulin resistance in obese type 2 diabetic patients exceeds that caused by obesity alone, and nonobese type 2 diabetic patients are also insulin resistant.3,5,101
All methods of assessing insulin resistance in vivo rely on measurement of the ability of a fixed dose or concentration of insulin to promote glucose disposal. Thus, a blunted decline in plasma glucose concentration after administration of intravenous insulin has been demonstrated in type 2 diabetes.102,103 Another approach has been to infuse insulin and glucose at fixed rates while endogenous insulin secretion is inhibited by a combination of epinephrine and propranolol or by somatostatin.102,104 With this method, the resulting steady-state plasma glucose level reflects the action of concomitantly infused insulin; the higher the steady-state plasma glucose, the greater is the degree of insulin resistance. Bergman and colleagues’ minimal model is yet another method of assessing in vivo insulin resistance.102 This method entails computer modeling of plasma glucose and insulin levels after an intravenous glucose bolus to generate an index of insulin sensitivity. With all these methods, type 2 diabetic subjects exhibit a significant decrease in insulin sensitivity compared with controls.3,102,105,106
More detailed studies of in vivo insulin resistance have been carried out with the euglycemic glucose clamp method.102 With this approach, insulin is infused at a constant rate, resulting in a given steady-state plasma insulin level, while plasma glucose is kept constant at a predetermined level by a feedback-controlled variable infusion of glucose. The insulin normally lowers the plasma glucose level by suppressing HGO and by stimulating tissue glucose uptake. During the insulin infusion, the amount of glucose that has to be infused to keep plasma glucose levels constant increases gradually until a steady state is reached. Under these steady-state conditions, the glucose disposal rate provides an excellent quantitative assessment of the biological effect of a particular steady-state insulin level. If a radioactive or stable isotope of glucose is also infused during the study, HGO during the clamp can be quantified. In type 2 diabetes, glucose disposal rates are 30% to 60% lower than those in normal subjects at any given insulin infusion rate. If several studies at different insulin levels are performed in a given subject, dose response curves for insulin-stimulated glucose disposal and suppression of HGO can be constructed. Patients with type 2 diabetes (obese and nonobese) exhibit both a rightward shift in their dose response curve (diminished sensitivity) and a marked decrease in their maximal rate of glucose disposal (decreased responsiveness) (Fig. 41-8). These changes tend to be more pronounced in obese diabetic patients, particularly at maximal glucose disposal rates (see Fig. 41-8). The insulin resistance of obese type 2 diabetic patients is significantly greater than that of nondiabetic obese subjects. Subjects with IGT tend to have a rightward shift in their dose response curves with normal maximal glucose disposal rates (see Fig. 41-8).
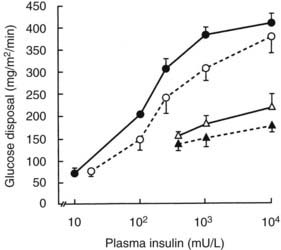
FIGURE 41-8. Mean insulin dose response curves for control subjects (closed circles), subjects with impaired glucose tolerance (open circles), and nonobese (open triangles) and obese (closed triangles) type 2 diabetic subjects. The group with impaired glucose tolerance has a rightward shift in the dose response curve without a change in maximal response (i.e., decreased sensitivity). Lean and obese patients with type 2 diabetes have both a rightward shift and a reduction in the response to a maximally stimulating concentration of insulin (i.e., decreased sensitivity and decreased insulin responsiveness).
(Data from Kolterman OG, Gray RS, Griffin J, et al: Receptor and post-receptor defects contribute to the insulin resistance in non-insulin dependent diabetes mellitus, J Clin Invest 68:957–969, 1981.)
Because skeletal muscle is responsible for the great majority of in vivo insulin-stimulated glucose uptake, this tissue must be the major site for resistance to insulin-stimulated glucose disposal. This conclusion is evidenced by the demonstration of insulin resistance in type 2 diabetic patients during forearm perfusion studies.107,108 Leg catheterization studies have shown that skeletal muscle accounts for ≈80% of whole-body insulin-mediated glucose uptake, and that leg skeletal muscle is markedly resistant to the ability of insulin to stimulate glucose uptake in type 2 diabetes.109–111 Thus, although other insulin target tissues display decreased insulin sensitivity, they do not account for a significant proportion of overall glucose uptake, and one can conclude that all measures of in vivo insulin action on glucose disposal largely assess the resistance of skeletal muscle to take up glucose under the influence of insulin.
PATHOPHYSIOLOGIC ABNORMALITIES IN INSULIN TARGET TISSUES
Skeletal Muscle
The major manifestation of skeletal muscle insulin resistance is decreased glucose disposal. Clearly, insulin action in skeletal muscle involves a cascade of events, and abnormalities anywhere along this sequence can lead to insulin resistance. These defects can involve abnormal coupling between insulin receptor complexes and the glucose transport system, decreased activity of the glucose transport system per se, or a variety of intracellular enzymatic defects located in various pathways of glucose metabolism.
Mechanisms of Skeletal Muscle Insulin Resistance
As the first step in insulin action, it is apparent that a decrease in cellular insulin receptors could lead to insulin resistance. However, this potential relationship is not as clear as it would seem because a maximal insulin effect is achieved at insulin concentrations that occupy a fraction of the surface receptors, giving rise to the concept of “spare” receptors. A maximal response of glucose transport in adipocytes and muscle is achieved with only 10% to 20% of the receptors occupied.112,113 Once the critical number of receptors needed to generate a maximal response is activated, additional increases in the prevailing insulin concentration lead to increases in receptor occupancy with no further increase in biological response, because a step (or steps) distal to the receptor is now rate limiting. The functional significance is that a decreased number of insulin receptors leads to a rightward shift in the insulin biological function dose response curve, with decreased responses at all submaximal insulin concentrations but a normal maximal response. A reduction in the maximal insulin response generally denotes the presence of a postbinding abnormality. In this context, the term postbinding defect includes abnormalities of insulin receptor function that affect its transmembrane signaling function, such as its kinase activity. A postreceptor defect refers to any abnormality in a step distal to the insulin receptor.
Early studies showed decreased insulin binding to circulating monocytes from obese and IGT subjects and from both obese and nonobese type 2 diabetic patients.114,115 This decreased binding was due to a decrease in insulin receptor number with no change in affinity. Similar results were subsequently obtained, when isolated adipocytes, hepatocytes, and skeletal muscle from obese subjects and patients with type 2 diabetes were used.116,117 The decrease in cellular insulin receptors in obesity and type 2 diabetes may well be secondary to hyperinsulinemia, inasmuch as elevated circulating insulin levels can downregulate receptor number.
A reduction in insulin receptor tyrosine kinase activity in type 2 diabetes generally has been found in patients with normal kinase activity in IGT.78,91,117 The receptor autophosphorylation/kinase defect appears to be generalized to all insulin target tissues and relatively specific for the hyperglycemic insulin-resistant state that is seen in type 2 diabetes.
Emerging evidence points to a role for IRS proteins in the development of insulin resistance. A defect in insulin-stimulated IRS-1 tyrosine phosphorylation is found in skeletal muscle from type 2 diabetic patients, although overall IRS-1 expression is unchanged.118 Serine/thr phosphorylation of IRS proteins is closely associated with reduction of signaling through IRS. Two potential mechanisms may underlie this phenomenon. First, serine phosphorylation may block the interaction of IRS-1 with its target proteins.119 Second, proteasomal-mediated degradation of IRS-1 may be increased.120 Several intermediary lipid metabolites and cytokines have been shown to activate a variety of serine/threonine kinases that induce serine phosphorylation of IRS-1. Serine kinases implicated include c-Jun NH2-terminal kinase (JNK),121 IκB kinase (IKK),122 PKC theta, S6K, and MTOR. It is interesting to note that this places IRS-1 at the intersection of a variety of intracellular pathways, including inflammation, endoplasmic reticulum (ER) stress, and nutrient sensing, all of which can activate serine/threonine kinases that may phosphorylate IRS-1.
IRS-2 is also important for insulin signaling and glucose homeostasis. In mice with disruption of the IRS-2 gene, profound defects in both insulin action (predominantly in the liver) and β cell function develop, progressing to diabetes.123
PI3K plays a key role in mediating the effects of insulin on glucose metabolism.124 Insulin-stimulated PI3K activity in skeletal muscle is reduced in both obese nondiabetic subjects125 and patients with type 2 diabetes.118 This reduction in PI3K activity correlates with the decrease in whole-body glucose disposal.118 An interesting twist on this relates to the regulatory subunits of PI3K. PI3K consists of a catalytic, 110 kDa subunit, as well as a family of regulatory subunits (p50, p55, and p85). If expression of the regulatory and catalytic subunits is unbalanced, with excess levels of the regulatory subunits, then monomeric p50/55/85 can bind through their SH2 domains to tyrosine phosphorylated substrates, such as IRS-1. When this happens, they can compete out binding of the dimeric p110.p85 PI3K complex, thus inhibiting PI3K signaling. Activated PI3K stimulates pyruvate dehydrogenase kinase (PDK1) in the plasma membrane, which then leads to activation of AKT and PKC λ/ζ. Both of these latter enzymes are upstream of GLUT4 translocation and are important regulators of glucose transport stimulation. Decreased insulin-induced AKT and PKC λ/ζ activation are widely described in insulin-resistant skeletal muscle from a variety of states.
Insulin-stimulated glucose transport in isolated muscle fibers and adipocytes from type 2 diabetic patients is markedly reduced at all insulin concentrations.10,126 What is the mechanism of this decrease? In adipocytes from type 2 diabetic subjects, decreased GLUT4 levels have been reported. In contrast, skeletal muscle GLUT4 mRNA and protein levels are normal in type 2 diabetes.127,128 Because the muscle of type 2 diabetic patients is not deficient in GLUT4 protein, it appears that the defect in insulin-stimulated glucose transport reflects a decrease in the ability of insulin to signal translocation of GLUT4 to the cell surface.
Indeed, clear evidence suggests that this is the case. For example, Kelley and coworkers used quantitative confocal laser scanning microscopy to examine insulin-stimulated recruitment of GLUT4 to the sarcolemma in muscle biopsies from patients with type 2 diabetes.98 In the basal state, sarcolemmal GLUT4 labeling was similar in diabetic and normal subjects, but in response to insulin, the increase in GLUT4 in type 2 diabetic subjects was only 25% of that in control subjects. A quantitatively similar defect in GLUT4 translocation was found in obese nondiabetic subjects. In both type 2 diabetic subjects and obese nondiabetic subjects, the defect in GLUT4 translocation was associated with marked impairment in insulin-stimulated muscle glucose transport as determined by positron emission tomography. Others using biochemical muscle subfractionation techniques have found a defect in GLUT4 translocation in patients with type 2 diabetes.99
Trafficking of GLUT4 involves a complex system analogous to synaptic vesicle movement, and an expanding list of proteins involved in the regulation of GLUT4 trafficking are being identified.100 Clearly, impaired GLUT4 translocation could be due to a defect in one or more of these GLUT4 vesicle-trafficking proteins, which is an area requiring intensive investigation.
Oxidative and Nonoxidative Glucose Metabolism in Skeletal Muscle
By performing indirect calorimetry during glucose clamp studies, one can determine the intracellular fate of glucose by measuring the percentage of glucose that is oxidized versus that which undergoes nonoxidative glucose metabolism (consisting of storage as glycogen plus glycolysis). The insulin concentrations necessary for half-maximal stimulation of glucose oxidation (≈50 mU/L in normal subjects) are lower than those required for stimulation of glucose uptake and storage as glycogen (≈100 mU/L).129 Thus, at low physiologic insulin levels, oxidative glucose disposal is quantitatively more important, but at higher insulin levels, nonoxidative glucose metabolism predominates.12 Defects in both oxidative and nonoxidative glucose metabolism exist in type 2 diabetes, although the decrease in nonoxidative metabolism is greater.9,36,111,130,131 Shulman and coworkers, using nuclear magnetic resonance (NMR) spectroscopy of the gastrocnemius muscle during an infusion of 13C-enriched glucose, showed that during a hyperinsulinemic hyperglycemic clamp study, nonoxidative glucose disposal is highly correlated with rates of skeletal muscle glycogen deposition.9 Moreover, a 50% reduction in the rate of muscle glycogen synthesis in type 2 diabetic patients was found, and defects in nonoxidative glucose metabolism and muscle glycogen synthesis correlated well with the decrease in whole-body glucose uptake.9
The reduced muscle glycogen synthesis rate in type 2 diabetes could be the result of a decrease in glucose transport, impaired glucose phosphorylation, or an abnormality in the glycogen synthetic pathway, and decreases in GLUT4 translocation, hexokinase II, and glycogen synthase have been reported. To identify the primary site of the intracellular block in glycogen synthesis, Rothman and coworkers used 31P-NMR during glucose clamp studies to measure glucose-6-phosphate concentrations in gastrocnemius muscle.132 They reasoned that a primary block in glycogen synthesis (e.g., resulting from decreased glycogen synthase) would lead to increased glucose-6-phosphate levels, whereas if the decreased flux of glucose to glycogen reflected impaired glucose transport and/or phosphorylation, then glucose-6-phosphate levels would be low. They found a lower steady-state glucose-6-phosphate concentration in type 2 diabetes, indicating that the reduced rate of glycogen synthesis was secondary to impaired glucose transport, hexokinase activity, or both. In additional muscle NMR studies, these investigators detected a very low intracellular free glucose concentration during hyperinsulinemic hyperglycemic clamp studies in both normal and type 2 diabetic patients.81 This finding strongly suggested that glucose transport is the rate-controlling step in insulin-stimulated muscle glycogen synthesis, indicating that decreased insulin-mediated glucose transport is the major defect in the muscle insulin resistance of type 2 diabetes.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


