Fig. 3.1
M2 tumor-associated macrophages (TAMs) support tumor growth and progression. Circulating monocytes recruited by tumor-derived chemoattractants develop into TAMs in the tumor microenvironment. TAMs generally show an M2-like phenotype. T helper (Th) 2 cytokines [interleukin (IL)-4 and IL-13] and IL-10 induce M2 polarization of TAMs. M2 TAMs produce tumor-promoting factors including immunosuppressive cytokines, proangiogenic factors, growth factors, and matrix-degrading enzymes [e.g., matrix metalloproteinases (MMPs)]. Stimulation with Toll-like receptor (TLR) ligands or cytokines influence the function of TAMs
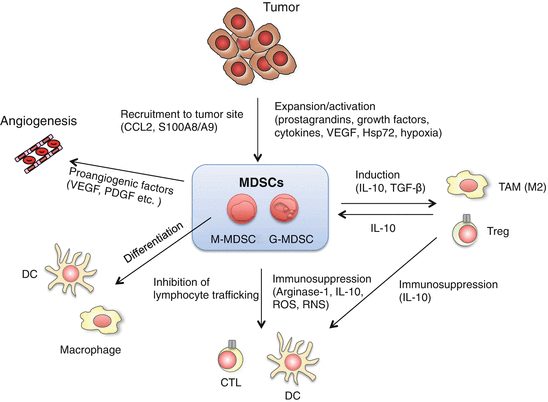
Fig. 3.2
Multiple roles of myeloid-derived suppressor cells (MDSCs) in tumor growth and progression. MDSCs consist of monocytic MDSCs (M-MDSCs or Mo-MDSCs) and granulocytic MDSCs (G-MDSCs), which are distinguished by the expression of Ly6G and Ly6C. M-MDSCs contain precursors of macrophages, dendritic cells (DCs) and G-MDSCs. MDSCs suppress anti-tumor responses through a variety of mechanisms. MDSCs suppress the activation and proliferation of T cells by production of arginase and reactive oxygen species (ROS), nitration of tyrosine residues of T cell receptors (TCRs), or deprivation of cysteine. MDSCs also produce interleukin (IL)-10 and tumor growth factor (TGF)-β, leading to induction of regulatory T cells (Tregs) and M2 tumor-associated macrophages (TAMs), and suppression of DC function. Proangiogenic factors derived from MDSCs promote tumor growth by regulating angiogenesis. Tumor-derived factors [prostaglandins, growth factors, cytokines, vascular endothelial growth factor (VEGF), and heat-shock protein (Hsp) 72) and hypoxia are involved in the development and the expansion of MDSCs. S100 calcium-binding protein (S100) A8/A9 and chemokine CC ligand (CCL) 2 recruit MDSCs to tumor sites. S100A8/A9 and Hsp72 modulate MDSC expansion and function through the activation of Toll-like receptor (TLR) 4 and TLR2, respectively. TLR signals and TLR signal-induced cytokines regulate MDSC development and expansion through intracellular signaling pathways
3.3.1 Tumor-Supportive Roles of Tumor-Associated Macrophages (TAMs)
TAMs frequently infiltrate multiple types of solid tumors in humans and mice (Solinas et al. 2009). Development and growth of mammary tumors are greatly reduced in macrophage colony-stimulating factor (M-CSF)-deficient op/op mice, which are deficient in macrophages due to the lack of M-CSF (Lin et al. 2001). Bisphosphonate treatment, which selectively depletes phagocytes including macrophages in vivo, decreases the number of TAMs and induces retardation of tumor growth (Rogers and Holen 2011). These studies suggest that TAMs are essential cells for tumor growth and progression. Both tumor cells and stromal cells in tumor tissue induce the recruitment of circulating monocytes in the peripheral blood by secreting a variety of chemoattractants such as chemokine CC ligand (CCL) 2, CCL5, colony-stimulating factor (CSF)-1 and chemokine CXC ligand (CXCL) 12. Monocytes differentiate into TAMs in response to the tumor microenvironment (Solinas et al. 2009). A recent report demonstrates that the spleen and bone marrow are reservoirs for TAM precursors in tumor-bearing hosts (Cortez-Retamozo et al. 2012). TAMs are also differentiated from tumor-infiltrated monocytic MDSCs (M-MDSCs or Mo-MDSCs) as described below.
Two distinct activation states of macrophages are referred to as classically/alternatively activated or M1/M2-polarized states (Mantovani et al. 2002; Biswas and Mantovani 2010). Classically activated M1 macrophages produce pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-12, IL-23, IL-6, and IL-1β, leading to induction of T helper (Th) 1-type immune response and high expression of inducible nitric oxide (NO) synthase (iNOS). In vitro experiments have shown that TNF-α and NO are involved in direct killing of certain types of tumor cells by M1 macrophages. In contrast, alternatively activated M2 macrophages are characterized by higher expression levels of IL-10, arginase-1, scavenger receptor (SR), and macrophage mannose receptor (MMR, CD206), and lower expression of pro-inflammatory cytokines such as IL-12, IL-23, IL-1β, and IL-6. M2 macrophages have poor antigen-presentation capability and immunosuppressive activity by secreting IL-10 and transforming growth factor (TGF)-β. They also produce molecules such as angiogenic factors and matrix metalloproteinases (MMPs), which are involved in tissue remodeling. M1/M2-polarization of macrophages is regulated by immune signaling (Hu et al. 2007; Lawrence and Natoli 2011). IFN-γ and IFN-β are potent stimulation factors of macrophages and induce M1-like macrophages through Janus kinase/signal transducer and activator of transcription (STAT) 1 activation (Toshchakov et al. 2002). M1 macrophages derived from granulocyte–M-CSF (GM-CSF)-treated human monocytes highly express IRF5 compared with M-CSF-induced M2 macrophages. Over-expression of IRF5 in M-CSF-induced M2 macrophages forces them into macrophages that express M1-specific cytokines, leading to both Th1 and Th17 cell development (Krausgruber et al. 2011). The Notch-recombination signal binding protein for immunoglobulin kappa J (RBPJ) pathway determines M1/M2 polarization by controlling the expression of M1-related genes in macrophages via transcription factor IRF8 (Xu et al. 2012). These results raise the possibility that TLR signaling may directly induce the expression of M1-related genes through IRF5 and IRF8 because these transcription factors participate in TLR signal-induced transcription of cytokine genes (Honda and Taniguchi 2006). In contrast, the jumonji domain-containing 3 (JmjD3)–IRF4 axis regulates M2 macrophage development (Satoh et al. 2010). JmjD3, a H3K27me demethylase, is induced in macrophages in response to the ligands of TLR2, TLR4, and TLR9, and inflammatory cytokines (De Santa et al. 2007). STAT6 and peroxisome proliferator-activated receptor (PPAR)-γ regulate M2 macrophage polarization (Charo 2007; Ishii et al. 2009). Suppressor of cytokine signaling (SOCS) 2 and SOCS3 are involved in M1 and M2 polarization, respectively, by regulating intracellular cytokine signaling (Spence et al. 2013). Furthermore, TLR signals induce chromatin remodeling to control the gene expression through IRF3 and other transcription factors (Foster et al. 2007), which may regulate the expression levels of the transcription factors involved in macrophage polarization. Most importantly, macrophage polarization is not stable (i.e., plasticity), and the manipulation of macrophage function could be achieved by regulating multiple intracellular signaling pathways (Biswas and Mantovani 2010; Lawrence and Natoli 2011; Sica and Mantovani 2012).
Gene expression analysis has demonstrated that TAMs largely display the phenotype that is typical of the M2-polarized macrophages (Mantovani et al. 2008) (Fig. 3.1). However, TAMs polarized into the M1 or M2 phenotype coexist in tumors but localize in different areas of the tumor. It is reported that infiltration of M2-polarized TAMs is correlated with poor prognosis in many types of cancers, including melanoma, colon cancer, and ovarian cancer (Lewis and Pollard 2006; Biswas and Mantovani 2010). Conversely, the density of M1 macrophages, defined as CD68+HLA-DR+, in the tumor is positively associated with the survival time of non-small cell lung cancer patients, whereas CD68+CD163+ M2 macrophages are not associated with patient survival (Ma et al. 2010). Thus, the balance of M1 versus M2 population is considered to affect tumor growth.
Macrophage polarization is affected by several factors in the tumor microenvironment. It has been demonstrated that tumor cell-derived TNF-α, TGF-β, prostaglandin E2 (PGE2), and hypoxia induce M2 polarization of macrophages (Lewis and Pollard 2006; Biswas and Mantovani 2010). Th2 cytokines such as IL-4 drive the development of M2-polarized TAMs (DeNardo et al. 2009). The recently identified transcription factors that drive macrophage polarization described above may explain a mechanism that regulates the development and polarization of TAMs.
Immune and non-immune mechanisms mediated by TAMs that affect tumor growth have been reported (Mantovani et al. 2008). M2 TAMs directly facilitate tumor growth by secreting a variety of growth factors for tumor cells. M2 TAM-secreted immunosuppressive cytokines such as IL-10 down-regulate the anti-tumor activity of cytotoxic T cells and NK cells. IL-10 may also promote tumor progression by potentiating Treg activity. Furthermore, M2 TAMs produce proangiogenic factors such as vascular endothelial growth factor (VEGF), chemokines such as CCL2, and CXCL8, and growth factors such as platelet-derived growth factor and epidermal growth factor, which stimulate formation of new blood vessels to supply nutrients that are essential for tumor growth (Lin and Pollard 2007). Furthermore, TAMs influence the efficacy of anticancer therapies including chemotherapy, tumor irradiation, vascular-targeted therapies, and antibody therapies (De Palma and Lewis 2013).
Increasing evidence shows that TAMs play critical roles in multiple stages of tumor growth and metastasis in which TLR signaling pathways are involved (Qian and Pollard 2010; Sica 2010). Macrophages are recruited into pre-metastatic organs by tumor-derived factors where TLR4-activated macrophages facilitate invasion and metastasis of tumor cells by secreting proteolytic enzymes such as MMP7 and MMP9 to destroy the extracellular matrix (Hiratsuka et al. 2006, 2008). Versican, an extracellular matrix protein, secreted by tumor cells induces TNF-α production by macrophages through activation of the TLR2 signaling pathway. This promotes tumor metastasis into lungs (Kim et al. 2009). Another study has suggested that activation of TLR4 signaling pathway on M2-polarized TAMs partially induces epithelial–mesenchymal transition (EMT) in pancreatic cancer through increased IL-10 production (Liu et al. 2013).
3.3.2 Expansion and Activation of Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs are a heterogeneous cell population that consists of myeloid progenitor cells and immature myeloid cells (iMCs). MDSCs are defined as CD11b+Gr1+ cells in mice and CD14−CD11b+CD33+HLA-DRneg/low in humans, and lack maturation markers of macrophages and DCs. MDSCs have potent immunosuppressive activity against both innate and adaptive immunity (Gabrilovich et al. 2012). Although CD11b+Gr1+ cells are normally present in healthy mice, they do not have immunosuppressive activity. Myeloid progenitor cells immediately differentiate into neutrophils, macrophages, or DCs in healthy mice. However, in tumor-bearing mice, the differentiation is blocked, which cause MDSC accumulation in spleen, blood, lymph nodes, and primary and metastasized solid tumors. They are also frequently detected in the peripheral blood of human cancer patients (Almand et al. 2001; Diaz-Montero et al. 2009; Ostrand-Rosenberg and Sinha 2009). In tumor tissues, MDSCs can be distinguished from TAMs by the expression of surface molecules. Gr1 is highly expressed on MDSCs but not TAMs, and F4/80 is expressed on TAMs but less on MDSCs. MDSCs are further characterized into two populations: Ly6G−Ly6Chigh M-MDSCs and Ly6G+Ly6Clow granulocytic MDSCs (G-MDSCs) or polymorphonuclear MDSCs (PMN-MDSCs) (Youn et al. 2008; Movahedi et al. 2008; Peranzoni et al. 2010). Although G-MDSCs are a major subset in tumors and peripheral blood, their suppressive activity is relatively low compared to that of M-MDSCs. Recently, it was revealed that M-MDSCs contain precursors of TAMs and G-MDSC (Youn et al. 2013). MDSCs are distributed throughout the body. Tumor-derived chemokines or chemoattractants such as CCL2, Bv8 (also known as prokineticin-2), and S100 calcium-binding protein (S100) A8/A9 recruit MDSCs from peripheral organs into tumor sites (Huang et al. 2007; Shojaei et al. 2007; Sawanobori et al. 2008; Sinha et al. 2008).
Inflammation-associated factors promote the expansion of MDSCs. Prostaglandins, stem-cell factor (SCF), M-CSF, GM-CSF, TNF-α, IL-1β, IL-6, PGE2, and VEGF promote MDSC expansion (Gabrilovich et al. 2012). Some of these molecules are under the control of TLR signals. In fact, TLR4 activation leads to suppressive activity of MDSCs through a MyD88/NF-κB-dependent mechanism. STAT3, which is activated by stimulation with some of these inflammatory molecules, is one of the key signaling molecules that regulates the expansion of MDSCs. STAT3 is frequently observed to be activated in tumor-infiltrating immune cells (Yu et al. 2007). MDSC expansion is not observed in STAT3 conditional knockout mice or STAT3-specific inhibitor-treated mice under tumor-bearing conditions, which results in an increase of T cell responses (Nefedova et al. 2005; Kortylewski et al. 2005). Constitutive activation of STAT3 leads to the production of S100A8 and S100A9 in iMCs, resulting in inhibition of their differentiation. Increased reactive oxygen species (ROS) concentration driven by S100A8/A9-induced nicotinamide adenosine dinucleotide phosphate (NADPH) activation results in differentiation of iMCs into MDSCs (Cheng et al. 2008). Hsp72-containing tumor-derived exosomes trigger STAT3 activation through TLR2/MyD88-dependent IL-6 production by autocrine mechanisms, which results in the induction of MDSC expansion (Chalmin et al. 2010). Another report shows that STAT3 or STAT5 down-regulates IRF8 to maintain MDSC development (Waight et al. 2013). S100A8 and S100A9 expression is up-regulated in many tumors, including gastric, lung, bladder, mammary, and colon cancer (Srikrishna 2011). Activated neutrophils and macrophages in tumor or necrotic tumor cells release the S100A8/A9 complex, which act as a chemoattractant for MDSCs. The S100A8/A9 complex promotes and amplifies inflammatory responses via direct binding to TLR4 (Ehrchen et al. 2009). Inflammation-induced TNF signaling drives the peripheral accumulation of MDSCs through TNF receptor (TNFR)-2, but not TNFR-1. TNF-α inhibits differentiation and enhances suppressive activity of iMCs during chronic inflammation, resulting in generation of MDSCs. TNF-α-induced S100A8 and S100A9 proteins and their corresponding receptor, receptor for advanced glycan endproducts (RAGE), augment MDSC-suppressive activity (Sade-Feldman et al. 2013). Activation of complement cascades accompanied by TLR-induced inflammation also regulates tumor growth by modulating MDSC function. Complement component C5a, a cleaved product of C5, is generated by inflammation in the tumor microenvironment and recruits MDSCs and enhances their suppressive function against CD8+ T cell proliferation, which contributes to tumor growth. Enhanced suppression is achieved by increased ROS and reactive nitrogen species (RNS) in M-MDSCs but not G-MDSCs, which results in reduced T cell responses against tumor cells (Markiewski et al. 2008). Another report shows that the signaling balance of paired immunoglobulin-like receptor (PIR) family members PIR-A and PIR-B are expressed on MDSCs and are important for the regulation of MDSC differentiation. MDSCs isolated from PIR-B-deficient Lilrb3−/− mice preferentially differentiate into an M1-like rather than M2-like immunosuppressive phenotype in Lewis lung cancer (LLC; also known as 3LL) tumor-bearing mice. LPS and IFN-γ stimulation enhances M1 polarization by suppressing STAT3 activation in the absence of PIR-B (Ma et al. 2011). Expansion of MDSCs are also regulated by other transcription factors (Condamine and Gabrilovich 2011; Sonda et al. 2011). Collectively, TLR signals are involved in MDSC expansion directly and indirectly.
3.3.3 Regulation of Immune Responses by MDSCs
MDSCs suppress anti-tumor T cell responses by several mechanisms (Fig. 3.2). The immunosuppressive activities of MDSCs are divided into four categories. First, inhibition of T cell proliferation is mediated by depleting nutrients in the microenvironment. MDSCs highly express arginase-1, which rapidly decreases the concentration of l-arginine in the microenvironment. Reduced level of l-arginine concentration causes a profound inhibition of T cell proliferation by the inability to up-regulate cyclin D3 and cyclin-dependent kinase 4 upon antigen stimulation, cytokine production, and expression of the CD3ζ chain of the T cell receptor (TCR) (Zea et al. 2005). MDSCs also inhibit T cell proliferation by sequestering cystine and limiting the availability of cysteine (Srivastava et al. 2010). Second, ROS and RNS produced by MDSCs modulate immune responses. MDSC-derived peroxynitrate inhibits T cell responses by inducing nitration of tyrosine residues in TCRs, resulting in an altered TCR/major histocompatibility complex (MHC) peptide recognition (Nagaraj et al. 2007). In parallel, peroxynitrite induces tumor cell resistance to CTLs by modifying MHC class I–antigen complex (Lu et al. 2011). Among two subsets, G-MDSCs suppress antigen-specific CD8+ T cells predominantly by producing ROS (Youn et al. 2008). Third, MDSCs modulate lymphocyte trafficking. CD62L (l-selectin) expression on naive CD4+ and CD8+ T cells are decreased by a disintegrin and metalloprotease 17 (ADAM17) on MDSCs, leading to the inhibition of recruitment to lymph nodes (Hanson et al. 2009). Fourth, MDSCs indirectly affect T cell activation by inducing immunomodulatory cells such as Foxp3+ Tregs and M2 TAMs. MDSC-derived TGF-β and IL-10 are required for Treg induction (Huang et al. 2006; Serafini et al. 2008). In addition to the differentiation of MDSCs into M2 macrophages in tumors, MDSC-derived IL-10 and cell–cell interaction promote M2 polarization of macrophages, as well as impair cytokine production and antigen presentation by DCs.
How MDSCs are implicated in the regulation of NK cell function is controversial. MDSCs inhibit NK cell cytotoxicity against tumor cells and IFN-γ production through direct cell–cell interaction. Membrane-bound TGF-β1 expressed on MDSCs inhibits NK cell cytotoxicity and IFN-γ production, and induces anergy of NK cells in a liver transplant model (Li et al. 2009). MDSCs also inhibit NK cell activation by blocking the expression of NK group 2D (NKG2D). However, another report suggests that the F4/80+ population of MDSCs express retinoic acid early inducible 1 (RAE-1), the ligand for NKG2D, activate NK cell cytotoxicity (Nausch et al. 2008).
These immunoregulatory functions of MDSCs are regulated by TLR and TLR-induced cytokine production. IFN-γ stimulation induces suppression of antigen-specific T cell responses by M-MDSCs, which requires STAT1 activation (Movahedi et al. 2008). STAT3 signaling is potentially activated by pro-inflammatory cytokines such as IL-6 and is implicated in arginase-1 expression in CD14+HLADR-/low MDSCs from head and neck cancer patients (Vasquez-Dunddel et al. 2013). Hypoxia-inducible factor (HIF)-1α activation enhances immunosuppressive function and differentiation of MDSCs in the tumor microenvironment (Corzo et al. 2010). LPS-induced TLR4 signal activates HIF-1α (Frede et al. 2006), suggesting that TLR4-triggered MDSC accumulation and development may be induced via HIF-1α-mediated transcriptional regulation. Activation of TLR as well as IL-1 receptor and receptor tyrosine kinases activate Gr1+CD11b+ MDSC-like cells to promote tumor inflammation and progression through phosphoinositide 3-kinase γ (Schmid et al. 2011).
3.4 Tumor-Associated Myeloid Cells as a Therapeutic Target of Cancer
Stromal cells in the tumor microenvironment are considered to be promising targets for cancer treatment (Quail and Joyce 2013). Recent reports suggest that regulation of immunosuppressive activity of tumor-associated myeloid cells could be useful for improving the efficacy of cancer immunotherapy. Therefore, strategies for elimination of tumor-associated myeloid cells or modulation of their function in tumor-bearing hosts are currently being investigated (Ugel et al. 2009; Talmadge and Gabrilovich 2013). Molecules that are responsible for the accumulation and immunosuppressive activity of MDSCs and TAMs could become therapeutic targets. There are several classes of inhibitors or reagents that can control the population or modulate the function of tumor-associated myeloid cells as described in other review (Ugel et al. 2009).
3.4.1 Re-education of Immunosuppressive Myeloid-Derived Cells Expanded in Cancer
Re-education of immunosuppressive cells toward cells with anti-tumor activity is an emerging concept of cancer therapy (Allavena and Mantovani 2012; Yang et al. 2013; Quail and Joyce 2013). It has been demonstrated that the conversion of TAMs and MDSCs into tumoricidal effector cells or ablation of their immunosuppressive activity results in tumor regression. Innate immune signaling induced by endogenous TLR ligands predominantly leads to expansion of TAMs and MDSCs with tumor-promoting activity, whereas manipulation of tumor-associated myeloid cell function could be achieved by using purified exogenous TLR ligands. Innate immune signaling on tumor-associated myeloid cells could be an attractive target for overcoming immune suppression induced by tumor and tumor-associated myeloid cells.
Purified TLR ligands have been studied in clinical trials for cancer immunotherapy (Galluzzi et al. 2012; Kaczanowska et al. 2013). It has been demonstrated that ligands of TLR2 [Pam3CSK4, Pam3CSK4, and bacillus Calmette-Guerin (BCG) and BCG cell wall skeleton (BCG-CWS)], TLR3 (poly I:C, poly I:C-LC, Ampligen, poly A:U), TLR4 (MPLA), TLR5 (flagellin), TLR7 and TLR8 (imiquimod), and TLR9 [CpG oligodeoxynucleotides (ODNs)] are capable of inhibiting tumor growth by modulating DC function in mouse models (Ahonen et al. 2004; Sfondrini et al. 2006; Huang et al. 2008; Seya et al. 2010). In spite of the presence of tumor-associated myeloid cells with immunosuppressive activity, these TLR ligands elicit innate immune responses against cancer. Therefore, innate immune signals seem to overcome the suppression by making an alteration of tumor-associated myeloid cell function. Recent reports have suggested that immunosuppressive TAMs and MDSCs could be converted into cells that have anti-tumor activity by manipulating signaling pathway induced by innate immune signaling (Figs. 3.3 and 3.4)
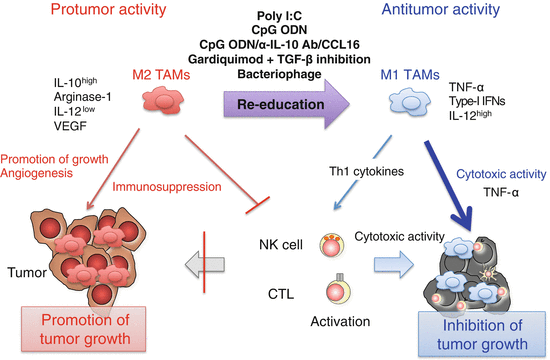
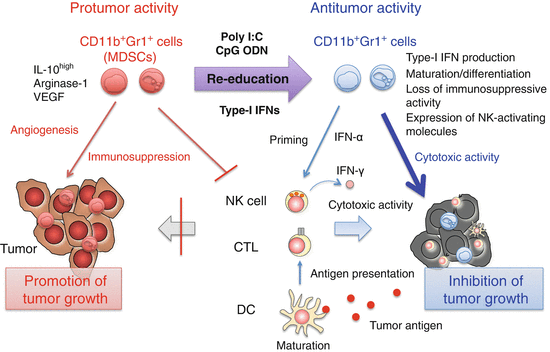
Fig. 3.3
Induction of M1-like tumor-associated macrophages (TAMs) by Toll-like receptor (TLR) activation. Poly I:C (TLR3), CpG oligodeoxynucleotide (ODN) (TLR9), or combined use of CpG ODNs, anti-interleukin (IL)-10 antibody, and chemokine CC ligand (CCL) 16-expressing vector, gardiquimod (TLR7), or bacteriophage alter the phenotype of TAMs from immunosuppressive M2 to anti-tumor M1 through activation of the intracellular TLR signaling pathways in TAMs. M1-like TAMs produce interferon (IFN)-β and pro-inflammatory cytokines including IL-12 and tumor necrosis factor (TNF)-α. TNF-α is responsible for direct killing of a certain type of tumor cells by M1-like TAMs. Other T helper (Th) 1 cytokines contribute to the activation of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells
Fig. 3.4
Modification of myeloid-derived suppressor cell (MDSC) function by innate immune signaling. Poly I:C [Toll-like receptor (TLR) 3 and melanoma differentiation-associated protein-5 (MDA5)], CpG oligodeoxynucleotide (ODN) (TLR9), and interferon (IFN)-α produced by CpG ODN-activated plasmacytoid dendritic cells (pDCs) induces maturation and activation of MDSCs. As a result, immunosuppression is abrogated and tumor growth is inhibited by restoring cytotoxic T lymphocyte (CTL) responses. Furthermore, MDSCs produce IFN-α and express natural killer (NK)-activating molecules in response to poly I:C, leading to IFN-γ production by NK cells
3.4.2 Modification of TAM Function by Innate Immune Signaling
M1 polarization of TAMs and tumor destruction are achieved by activating innate immune signaling. Bacteriophages induce secretion of M1-related pro-inflammatory cytokines and increased expression of molecules involved in antigen presentation and co-stimulation in TAMs through the TLR/MyD88 pathway (Eriksson et al. 2009). Another report shows that Notch signaling determines M1/M2 polarization in tumor-infiltrating macrophages (Wang et al. 2010). Forced activation of Notch in macrophages promotes M1 polarization while repressing M2 polarization through RBPJ. TLR4 signal-induced NF-κB and the IL-1 receptor-associated kinase 2 (IRAK2)/ MAPK-interacting kinase 1 (Mnk1) pathway cooperatively activate RBPJ signaling. Notch/RBPJ augments TLR4-induced M1-related genes via IRF8 expression in macrophages (Xu et al. 2012). Therefore, TLR4 ligands may affect macrophage polarization in tumors by regulating the Notch/RBPJ signaling pathway. TLR signals mostly activate NF-κB to promote expression of pro-inflammatory cytokines. However, NF-κB activation induced by IL-1β/MyD88 signaling is involved in M2 polarization of TAMs and targeting of NF-κB signaling can re-educate TAMs to become M1 macrophages (Hagemann et al. 2008). When NF-κB signaling is inhibited by using dominant negative form of IκB kinase β (IKKβ), TAMs show an M1-polarized phenotype represented by IL-12high and MHC Ihigh, but IL-10low and arginase-1low and anti-tumor activity. We have recently demonstrated that poly I:C administration into tumor-bearing mice leads to re-education of TAMs, resulting in retardation of tumor growth (Shime et al. 2012). In mice implanted with 3LL lung cancer cells, poly I:C injection rapidly (within 1 h) up-regulates TNF-α production in tumor by F4/80+ TAMs but not CD11b+Gr1+ MDSCs. It results in retardation of tumor growth by TNF-α-induced direct killing of 3LL tumor cells and hemorrhagic necrosis of the tumor. Poly I:C treatment up-regulates expression of M1-related genes such as I FN-β, IL-12, IL-1β, IL-6, and Cxcll1, while the expression of M2-related genes such as Arg1 is not affected. These responses require the activation of the TLR3/TICAM-1 (TRIF) signaling pathway, which is mostly essential for poly I:C-induced CD8+DC activation to induce cytotoxicity of CTLs and NK cells (Akazawa et al. 2007; Azuma et al. 2012; Seya et al. 2012). Thus, the TLR3/TICAM-1 signaling pathway induces re-education of TAMs as well as DC activation. A recent study demonstrated that macrophages stimulated with tumor-derived versican produce TNF-α, which promotes tumor metastasis to the lungs (Kim et al. 2009). Besides, MDSC accumulation in tumor-bearing mice is induced by TNF-α signaling (Zhao et al. 2012). These study suggest that TNF-α has both positive and negative effect on tumor growth and metastasis by regulating TAMs and MDSCs. These divergent effects of TNF-α in tumor regulation may be explained by the difference in the local concentration of TNF-α in the tumor between tumor development and treatment with TLR ligand. TAMs accumulate in 3LL tumors, where local TNF-α concentration transiently increases in response to poly I:C stimulation. Anti-tumor activity of TNF-α has been demonstrated by using a high dose of exogenous TNF-α or forced expression of TNF-α in tumor cell lines (Blankenstein et al. 1991; Zhao et al. 2007). Therefore, concentration of TNF-α derived from accumulated TAMs seems to be a critical determinant for growth of certain types of tumor cells.
Combined use of TLR ligand and other reagents modifying intracellular signaling in TAMs is demonstrated to be effective for inhibiting tumor growth mediated by tumor-associated myeloid cells. Local injection of CpG ODN and adenovirus encoding CCL16 chemokine into tumor, combined with systemic IL-10 receptor (IL-10R) antibody, effectively induces tumor eradication. In this case, resident tumor-infiltrating macrophages with M2-like phenotype are switched to macrophages with M1-like phenotype. Consequently, tumor-infiltrating DCs are matured and secrete pro-inflammatory cytokines to induce adaptive immune responses for tumor rejection (Guiducci et al. 2005). TLR7 ligand in combination with blocking reagent for TGF-β signaling induces the conversion of TAMs in their phenotype from M2 to M1. As a result, tumor apoptosis is increased and the number of CD4+, CD8+ , and CD19+ cells as well as neutrophils infiltrated into tumor is elevated (Peng et al. 2013).
3.4.3 Modification of MDSC Function by Innate Immune Signaling
TLR ligands alter MDSCs into cells with anti-tumor activity directly and indirectly. CpG ODN directly modifies MDSC function. Shirota et al. have reported that intratumoral injection of CpG ODN stimulates MDSCs to lose their suppressive activity on T cell proliferation, produce Th1 cytokines, and differentiate into macrophages with tumoricidal capability (Shirota et al. 2012). Other reports suggest that type-I IFNs produced in response to TLR stimulation modify MDSC function. Type-I IFNs are critical cytokines required for efficient anti-tumor immune responses elicited by TLR3, TLR7, and TLR9 signaling pathways. It has been demonstrated that type-I IFNs modulate the function of not only tissue-resident DCs and macrophages but also tumor-associated myeloid cells. Zoglmeier et al. have reported that CpG ODN treatment decreases the suppressive activity of MDSCs in mice bearing C26 tumors and CEA424-Tag mice bearing autochthonous gastric tumors (Zoglmeier et al. 2011). CpG induces maturation of MDSCs through plasmacytoid DC (pDC)-mediated type-I IFN production, which results in reduction of suppressive activity of MDSCs on T cell proliferation. Poly I:C also has similar activity on MDSCs via type-I IFN production, probably by hematopoietic and non-hematopoietic cells that express TLR3 and/or melanoma differentiation-associated protein-5 (MDA5) (McCartney et al. 2009). MDSCs activated by in vivo poly I:C treatment also produce IFN-α, implying the existence of an autocrine mechanism for MDSC activation (Shime et al. 2014). We have revealed that poly I:C treatment also induces maturation and activation of CD11b+Gr1+ MDSC-like cells to induce partial activation of NK cells (i.e., up-regulation of IFN-γ production, but not cytotoxicity) (Shime et al. 2014). Poly I:C treatment induces activation of MDSCs to produce IFN-α and express several surface molecules that are known to be involved in matured DC-mediated NK cell activation, such as RAE-1, IL-15, CD70, CD155 [poliovirus receptor (PVR)], CD150 [signaling lymphocyte-activation molecule (SLAM)] and IRF-3-dependent NK-activating molecule (INAM) (Newman and Riley 2007; Ebihara et al. 2010). In this pathway, IFN-α/-β receptor (IFNAR) signaling triggered by IFN-α on NK cells is critical for NK cell IFN-γ production induced by MDSCs from poly I:C-treated tumor-bearing mice. IFN-α produced through activation of the Mitochondrial Antiviral Signaling Protein (MAVS) (also known as IPS-1, VISA, and Cardif) pathway is responsible for both MDSC activation and NK cell priming by MDSCs. Therefore, MDSCs could be re-educated to have anti-tumor activity through the activation of the poly I:C-triggered MAVS signaling pathway. These reports suggest that type-I IFNs are critical factors for re-education of MDSCs.
Therapeutic use of TLR ligands combined with other agents has been proposed (Vanneman and Dranoff 2012). However, it should be noted that inappropriate use of TLR ligand as an adjuvant may induce undesirable effects on tumor growth. Imiquimod, a TLR7 ligand, significantly increases the levels of MDSCs and Tregs in mice immunized with self tumor antigen such as insulin-like growth factor-binding protein-2 (IGFBP-2) (Dang et al. 2012). Moreover, imiquimod reduces anti-tumor immunity induced by GM-CSF treatment through the expansion of MDSCs and Tregs. Although TLR ligands effectively induce anti-tumor immunity and tumor regression, it should be taken into consideration that they may have undesirable effects when used in combination therapy with other reagents.
3.5 Concluding Remarks
TAMs and MDSCs are proposed to be a target of cancer immunotherapy because they frequently accumulate in solid tumors and have critical roles in tumor growth and progression. Furthermore, they display a high degree of plasticity in their phenotype. Recent results have highlighted that TLR signaling pathways have important roles for switching between immunosuppressive phenotype and anti-tumor phenotype of TAMs and MDSCs.
Adjuvant immunotherapy using purified TLR ligands or TLR agonists seems to be a promising treatment for cancer by inducing DC-mediated anti-tumor responses. Recent reports suggest that TAMs and MDSCs play important roles in adjuvant therapy. However, it is true that TLR signals such as TLR2 and TLR4 contribute to promote tumor growth by inducing immunosuppressive activity of TAMs and MDSCs. Timing of administration and selection of TLR ligands may determine the outcome of adjuvant immunotherapy for cancer because the tumor microenvironment continuously changes during the course of cancer progression where the population and the function of TAMs and MDSCs are varied. Recent studies show that poly I:C is capable of inducing not only DC-mediated anticancer immune responses that lead to the activation of NK cells and CTLs, but also anti-tumor activity of TAMs and MDSCs. Therefore, poly I:C-induced TLR3/TICAM-1 and MDA5/MAVS pathways are promising targets of cancer treatment. Further basic studies to clarify the mechanisms of anti-tumor and pro-tumor effects induced by innate immune signaling on tumor-associated myeloid cells are still required to establish adjuvant therapy for cancer.
Acknowledgments
We apologize to the authors whose work we could not cite or illustrate in the figures because of space limitations. This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, and Culture (MEXT), “the Carcinogenic Spiral” (a MEXT Grant-in-Project), the Ministry of Health, Labor, and Welfare of Japan, the Takeda Foundation, the Akiyama Foundation, and the Kato Memorial Bioscience Foundation.
References
Ahonen CL, Doxsee CL, McGurran SM et al (2004) Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med 199:775–784. doi:10.1084/jem.20031591 PubMedCentralPubMed
Related posts:
 A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
 PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
 Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
 Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
 Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


