Fig. 2.1
Schematic drawing of the inflammatory microenvironment in gastrointestinal tumors. The cyclo-oxygenase (COX)-2/prostaglandin (PG) E2 pathway is important for the development and maintenance of the inflammatory microenvironment, including bone marrow-derived cells (BMDCs). PGE2 signaling also accelerates angiogenesis and cell survival as well as induces DNA methylation. In the activated macrophages, nuclear factor (NF)-κB induces the expression of growth factors and cytokines. The activation of NF-κB and Stat3 in tumor cells suppresses the apoptosis of tumor cells. NF-κB activation also induces the acquisition of stem cell properties (Modified from Oshima and Oshima 2012 with permission from Springer)
Importantly, disruption of the COX-2 gene or treatment with COX-2-selective inhibitors resulted in significant suppression of the polyp formation in Apc Δ716 mice (Oshima et al. 1996, 2001). Consistently, disruption of the mPGES-1 gene also suppressed intestinal polyposis in other Apc gene mutant mice (Nakanishi et al. 2008). Moreover, blocking PGE2 signaling through the EP2 receptor led to significant suppression of polyp formation in Apc Δ716 mice (Sonoshita et al. 2001). These results clearly indicate that the induction of COX-2/PGE2 signaling in the microenvironment is required for tumor development.
Many studies have shown the functions of PGE2 in tumorigenesis; i.e., angiogenesis through the induction of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) (Seno et al. 2002), and suppression of apoptosis by the activation of peroxisome proliferator-activated receptor (PPAR)-δ (Wang et al. 2004). Moreover, PGE2 signaling induces DNA methylation that silences tumor suppressor and DNA repair genes (Xia et al. 2012) (Fig. 2.1). However, the main mechanism by which PGE2 promotes cancer is still unclear. We have constructed a new mouse model, K19-C2mE mice, which express COX-2 and mPGES-1 simultaneously in the stomach, which induces chronic PGE2-associated inflammation in the gastric mucosa (Oshima et al. 2004) (Fig. 2.2). Therefore, one of the important mechanisms by which the COX-2/PGE2 pathway is involved in tumorigenesis is by generating an inflammatory microenvironment.
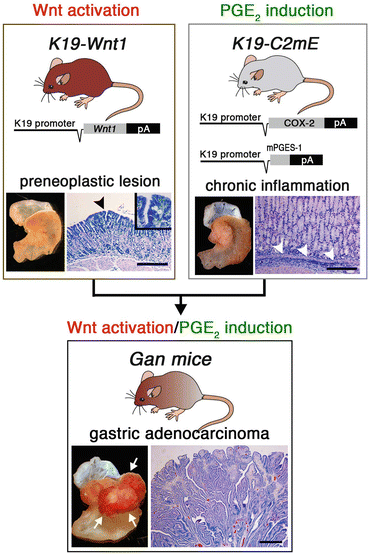
Fig. 2.2
Transgenic mouse models used to examine gastric tumorigenesis. Transgenic vector constructs and representative macroscopic and microscopic photographs of the stomach are shown for each line. Gan mice are compound K19-Wnt1 and K19-C2mE transgenic mice. The arrowhead in the K19-Wnt1 mouse stomach indicates a preneoplastic lesion. The arrowheads in the K19-C2mE mouse stomach indicate inflammatory infiltration. The arrows in the Gan mouse indicate gastric tumors. Bars indicate 100 μm (Modified from Oshima et al. 2009 with permission from Wiley)
2.4 Nuclear Factor (NF)-κB Signaling and Inflammation-Associated Colon Cancer
In the tissue exposed to chronic inflammation, lipid mediators such as PGE2, chemokines, and cytokines are induced, resulting in the recruitment of bone marrow-derived cells (BMDCs) such as macrophages, and activation of these cells leads to the development of a cytokine network. Several transcription factors, such as nuclear factor (NF)-κB and Stat3, are activated in the inflammatory lesions. NF-κB is activated by TNF-α signaling and Stat3 is activated by interleukin (IL)-6/IL-11, and these transcription factors both play a role in tumor promotion (Oshima and Oshima 2012). The role of Stat3 induction by IL-6 or IL-11 in intestinal tumorigenesis has been shown in genetic studies (Bollrath et al. 2009; Grivennikov et al. 2009; Putoczki et al. 2013). In this chapter, we discuss the role of NF-κB in the promotion of colon cancer development.
The treatment of mice with chemical mutagen azoxymethane (AOM) induces β-catenin gene mutations, and treatment with dextran sodium sulfate (DSS) in the drinking water induces ulcerative colitis. It is well-established in a mouse model that colitis-associated colon cancer can be induced by treatment with a combination of AOM and DSS. Genetic inactivation of NF-κB in the bone marrow cells of AOM/DSS-treated mice resulted in a decrease in the growth factor expression by stromal cells and suppression of colon tumor development (Greten et al. 2004). Notably, inactivation of NF-κB in intestinal epithelial cells also suppressed colon tumor development by leading to decreased expression of anti-apoptotic factors. These results indicate that inflammatory responses promote tumorigenesis through the activation of NF-κB in both BMDCs as well as tumor epithelial cells by inducing the expression of growth factors and suppressing apoptosis (Fig. 2.1).
Recently, it has been reported that NF-κB is important for the stem cell phenotype. Normal intestinal epithelial cells on villi are terminally differentiated and never proliferate. When Wnt signaling is activated in mouse villous epithelial cells by conditional mutagenesis in the β-catenin gene, the cells do not proliferate, indicating that Wnt activation cannot reset the terminal differentiation. However, if NF-κB is activated together with Wnt signaling, the epithelial cells acquire stem cell properties and start proliferating (Schwitalla et al. 2013a). These results suggest that inflammatory responses contribute to the development or maintenance of an undifferentiated status or stemness of cancer cells through NF-κB activation.
Although infection contributes to the development of an inflammatory microenvironment, it has not been elucidated how chronic inflammation is induced in non-infectious cancer tissues. Recent results have suggested the possibility that there may be oncogene-induced inflammation in tumor tissues. A loss or mutation of the p53 gene accelerates NF-κB activation in the microenvironment, resulting in an acceleration of colitis-associated tumor development in a mouse model (Schwitalla et al. 2013b; Cooks et al. 2013). Moreover, KRAS mutation causes TANK-binding kinase 1 (TBK1)-dependent NF-κB activation, which is required for the survival of cancer cells (Barbie et al. 2009). Accordingly, genetic alterations in oncogenes or tumor suppressor genes promote tumorigenesis not only by its intrinsic oncogenic activations, but also by inducing inflammatory responses.
2.5 Gan Mice, a Model of Inflammation-Associated Gastric Cancer
Helicobacter pylori infection is strongly associated with the development of gastric cancer. This infection induces activation of the COX-2/PGE2 pathway, which plays a role in infection-associated gastritis. We have constructed transgenic K19-Wnt1 mice that express Wnt1, a ligand for canonical Wnt signaling, specifically in the gastric epithelial cells (Oshima et al. 2006, 2009). These K19-Wnt1 mice develop small preneoplastic lesions in the gastric mucosa, but they do not develop tumors (Fig. 2.2). We crossed K19-Wnt1 mice and K19-C2mE mice to induce activation of both Wnt signaling and the COX-2/PGE2 pathway simultaneously in the stomach (Oshima et al. 2006, 2009). Importantly, these double transgenic mice, Gan mice, develop large gastric tumors with a 100 % incidence, indicating that the cooperation of Wnt signaling and the COX-2/PGE2 pathway causes gastric tumor development (Fig. 2.2). Moreover, the gene expression profiles of Gan mouse tumor tissues are similar to those of human intestinal-type gastric cancer (Itadani et al. 2009). Accordingly, it can be concluded that Gan mice develop gastric cancer by the same mechanism as human gastric cancer—the induction of oncogenic activation and inflammatory responses—and the expression profiles also reflect those of human gastric cancer.
2.6 Tumor Necrosis Factor (TNF)-α Signaling in Gan Mouse Gastric Tumorigenesis
In the gastric tumors of Gan mice, macrophages infiltrate into the tumor stroma, and they are activated to express proinflammatory cytokines, chemokines, growth factors, and proteases (Oshima et al. 2011a). We found that the expression of epidermal growth factor receptor (EGFR) ligands, including amphiregulin and epiregulin, and ADAM (A Disintegrin And Metalloproteinase) family proteases, is upregulated in Gan mouse tumors by an inflammation-dependent mechanism (Oshima et al. 2011b). The ADAM family proteases activate EGFR ligands by exodomain shedding, resulting in the activation of EGFR signaling, which may be one of the mechanisms by which inflammation leads to tumor promotion.
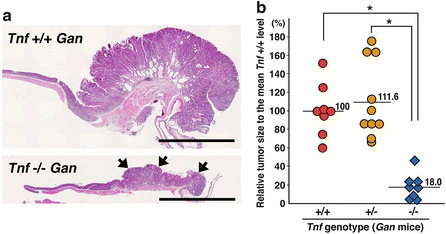
Among the various proinflammatory cytokines, we have been focusing on TNF-α, because it has been shown that disruption of TNF-α or the TNR-α receptor gene resulted in suppression of chemical carcinogen-induced tumor development in mouse models (Oshima et al. 2013). To examine the role of TNF-α in gastric tumorigenesis, we crossed Gan mice with TNF-α gene (Tnf) knockout mice to generate Tnf −/− Gan mice (Oshima et al. 2013). Notably, gastric tumorigenesis was significantly suppressed in Tnf −/− Gan mice, and the tumor volume decreased to 18 % of the size of the tumors in the Tnf wild-type Gan mice (Fig. 2.3). Notably, COX-2 and mPGES-1 are constitutively expressed in the Tnf −/− Gan mouse stomach because they are expressed by exogenous promoters; thus, it is possible that TNF-α signaling is required for tumor promotion even if the PGE2 pathway is activated.
 Development of Immunotherapy for Hepatocellular Carcinoma
Development of Glypican-3-Targeted Cancer Immunotherapy
Emerging Roles of an Innate Immune Regulator TAPE in the Toll-Like Receptor and RIG-I-Like Receptor Pathways
Host Factors Involved in the Propagation and Pathogenesis of Hepatitis C Virus
Immunological Regulation of Human Cancer Stem Cells/Cancer-Initiating Cells
Development of Personalized Combination Cancer Immunotherapy Based on the Patients’ Immune Status
Development of Immunotherapy for Hepatocellular Carcinoma
Development of Glypican-3-Targeted Cancer Immunotherapy
Emerging Roles of an Innate Immune Regulator TAPE in the Toll-Like Receptor and RIG-I-Like Receptor Pathways
Host Factors Involved in the Propagation and Pathogenesis of Hepatitis C Virus
Immunological Regulation of Human Cancer Stem Cells/Cancer-Initiating Cells
Development of Personalized Combination Cancer Immunotherapy Based on the Patients’ Immune Status




Related posts:
Development of Immunotherapy for Hepatocellular Carcinoma
Development of Glypican-3-Targeted Cancer Immunotherapy
Emerging Roles of an Innate Immune Regulator TAPE in the Toll-Like Receptor and RIG-I-Like Receptor Pathways
Host Factors Involved in the Propagation and Pathogenesis of Hepatitis C Virus
Immunological Regulation of Human Cancer Stem Cells/Cancer-Initiating Cells
Development of Personalized Combination Cancer Immunotherapy Based on the Patients’ Immune Status
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
